
- Background
- Training and Features
- RNA-binding ability prediction
- RNA-binding regions prediction
- Datasets
catRAPID signature Documentation
catRAPID signature predicts ribonucleoproteins and RNA-binding regions using physico-chemical properties instead of sequence similarity searches. The method is an extension of our previously published predictors catRAPID that predicts protein-RNA interactions, and CleverMachine that classify proteins for their physico-chemical features.
Background
RNA-binding proteins (RBPs) use RNA-binding domains (RDs) to recognize target RNAs and to regulate co- and post-transcriptional processes in eukaryotic cells. Examples of classical RDs include RNA-recognition motif (RRM), double-stranded RNA-binding domain (dsRRM), K-homology, RGG box and the Pumilio/FBF (PUM) domain. In addition to RDs, recent experimental studies (Baltz et al., 2012; Castello et al.; 2012; Kwon et al., 2013) indicate that a number of non-classical (ncRD) or putative RNA-binding domains (for details about these categories see Castello et al.; 2012 and Kwon et al., 2013) could interact with transcripts.
Training and Features
To characterize RNA-binding proteins we exploit 80 physico-chemical properties such as hydrophobicity, secondary structure, disorder, burial, aggregation, membrane and nucleic acid-binding propensities (Klus et al., 2014; Bellucci et al., 2011). For each property, we generate a profile assigning specific values to each amino acid in the sequence. Using profiles we calculate the correlations between RD and protein regions. In the training phase we investigated 950 human RBPs and 125 annotated PFAM domains for each physico-chemical property [sequence similarity reduced to 30% using CD-HIT (Li et al., 2006)]. Analysis of proteins containing annotated RDs (blue line), ncRDs (red line) and putative RDs (green line) reveals that a subset (list, sequences) is sufficient to differentiate more than 90% of the training set with respect to the proteome fraction that does not bind to RNA (see graph below and Dataset section).
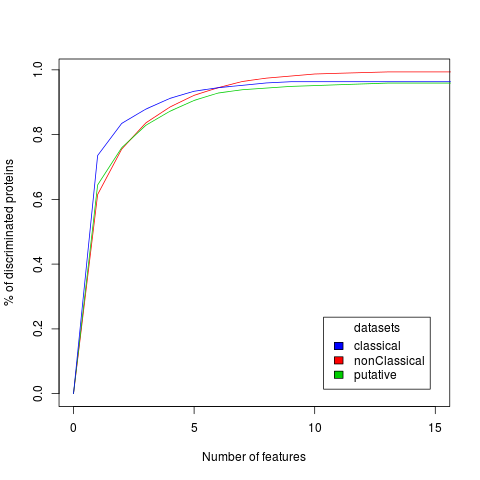
RNA-binding ability prediction
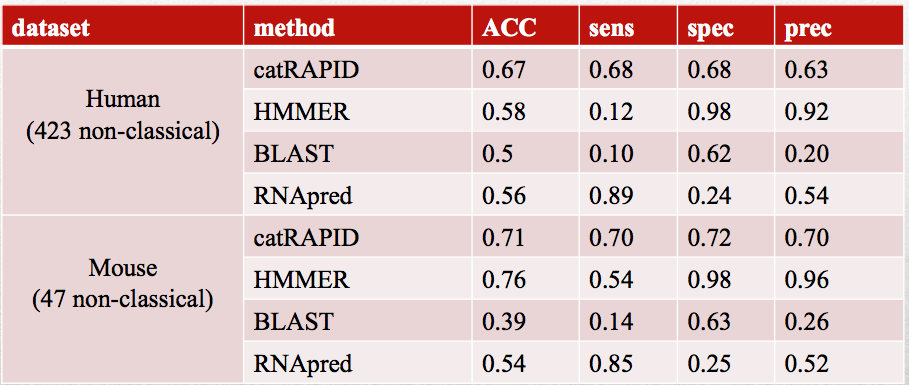
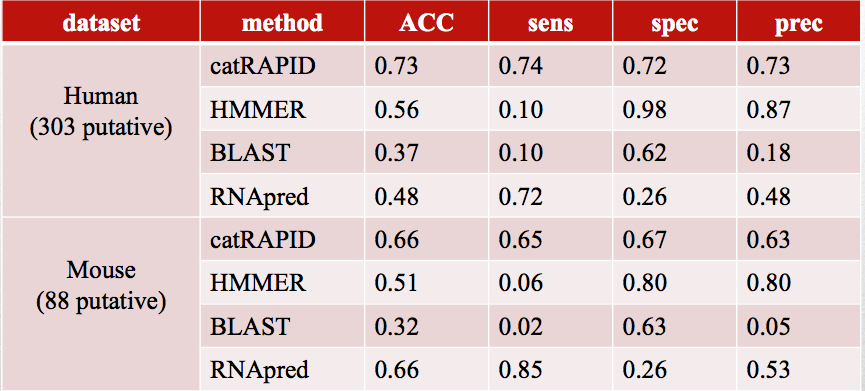
A machine learning approach (Pedregosa et al., 2011) was employed on the correlation values of the preselected domains and physico-chemical properties to predict the RNA-binding propensity of a protein. Training on the human dataset and performing cross-organism validation on mouse proteins, we observed high confidence predictions for newly discovered RNA-binding proteins. Performances with other methods are reported in the tables below. Comparisons have been done using: RNApred a method predicting the RNA-binding ability of a protein, HMMER a classical and well established domain-searching tool and BLAST a sequence similarity search tool.
Performances on mouse newly discovered RNA-binding proteins
Performances on different species (GO-term enrichment)
To assess the performance of catRAPID signature on other species a GO-term enrichment analysis has been carried out.
Seven reference proteomes have been downloaded from UniProt covering the kingdom of eukaryotes, fungi, bacteria and plants: Mus musculus (UP000000589), Homo Sapiens (UP000005640), Candida albicans (UP000001429), Saccharomyces cerevisiae (UP000002311),
Mycoplasma pulmonis (UP000000528), Escherichia coli (UP000000625), Arabidopsis thaliana (UP000006548) and Aspergillus oryza (UP000006564).
GO annotations files have been downloaded from ftp.ebi.ac.uk.
The proteins associated with annotation ribonucleoprotein complex (GO:0030529) are 1009 out of 43678 in
H. sapiens, 688/45185 in M. musculus, 818/35217 in A. thaliana, 359/12072 in A. oryza, 168/5742 in C. albicans, 608/6721 in S. cerevisiae, 63/4305 in E. coli and 52/778 in
M. pulmonis.
Using a cut-off value of 0.01, we found strong enrichments in all species.
The following barplot shows the significance (-log of p-value; y-axis) of the enrichment for different species (x-axis). The significance threshold of 0.01 is shown as dotted line.
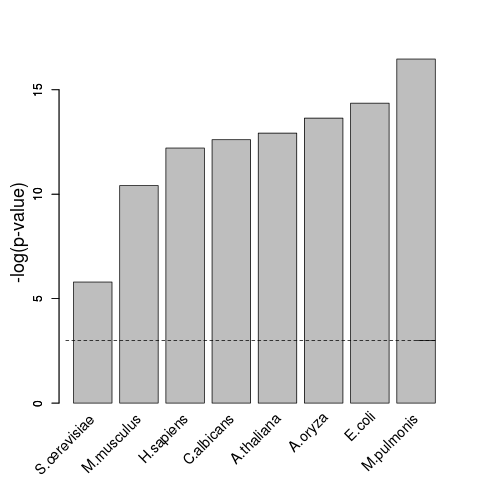 The analysis confirmed that catRAPID signature can reliably detect proteins associated with RNA-binding activity, not only in mammalian but also in other species and across different kingdoms.
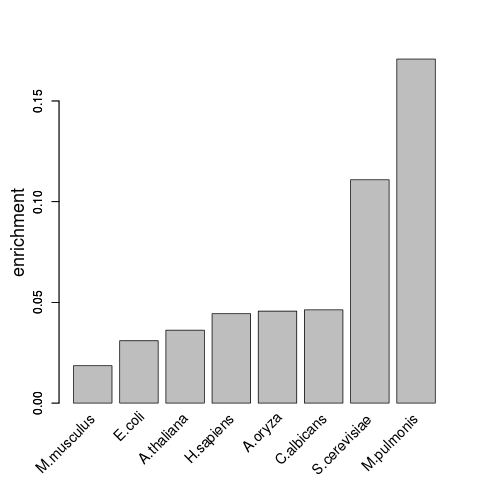
The following plot shows the enrichment (y-axis) of the GO-term ribonucleoprotein complex in different proteomes (x-axis).
The analysis confirmed that catRAPID signature can reliably detect proteins associated with RNA-binding activity, not only in mammalian but also in other species and across different kingdoms.
The following plot shows the enrichment (y-axis) of the GO-term ribonucleoprotein complex in different proteomes (x-axis).
RNA-binding regions prediction
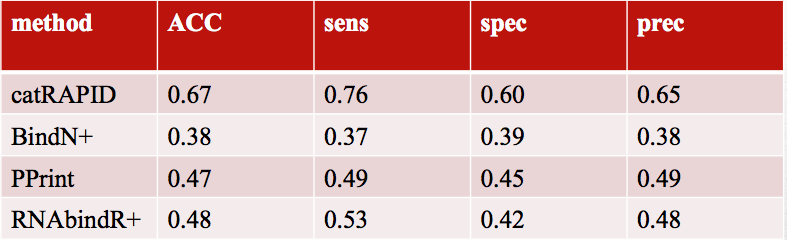
The set of preselected domains and physico-chemical properties is also used for identification of RNA-binding regions. The training has been carried out on 1115 human RDs comparing with 1115 same-length regions extracted from protein regions that do not bind RNA. Using 102 mouse ncRDs (vs. 102 non-binding regions) for independent validation the overall performances confirm the strength of our method to identify RBPs using physico-chemical sequence properties instead of using similarity searches. Performances with other methods are reported in the table below. Comparisons have been done using three methods that predict the RNA-binding residues in a protein: BindN+, PPRInt and RNABindRPlus
Performances on mouse RNA-binding regions
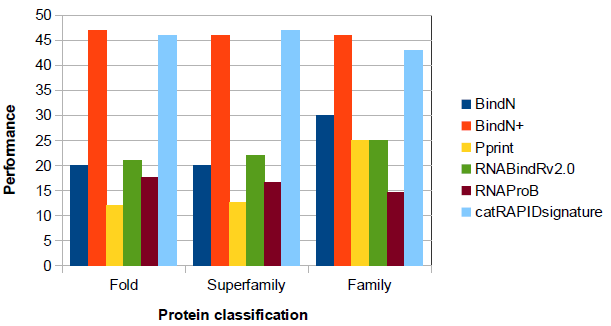
Performance on structural datasets for several classes of proteins
The following barplot shows the performances of catRAPID signature and different binding-residue predictors on a structural dataset (3.5 Å), that has been divided in different protein classes: Fold, Superfamily and Family (Nagarajan et al., 2014).
Datasets
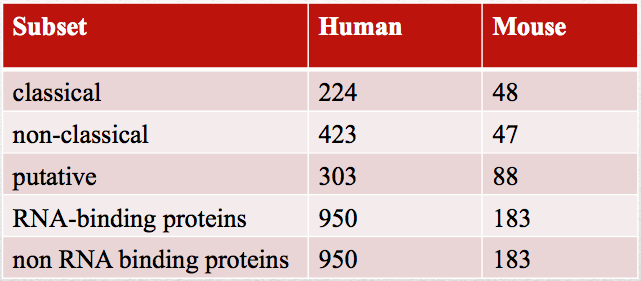
RNA-binding proteins were taken from studies on HeLA (Castello et al., 2012), HEK293 (Baltz et al., 2012) and mouse embryonic stem cells mESC (Kwon et al., 2013). [Dataset IDs and attributed categories: HeLa, HEK293, mESC]. Experimentally-validated negatives are taken from HeLa lysate (Castello et al.; 2012). CD-HIT (Li et al., 2006) was employed to reduce sequence similarity to 30% (respective sequences in FASTA format can be found here HeLa, HEK293, mESC).
Dataset composition
Implementation
To identify the best classification method for each task, we compared the performances of different machine learning algorithms. Here we tested the classification abilities of: Random Forest, Support Vector Machine, Nearest Neighbor, Extremely Randomized Trees and AdaBoost. The machine learning algorithms are taken from scikit-learn package (Pedregosa et al., 2011). We chose the best performing algorithm in a 10-fold cross-validation by maximizing the AUC.
catRAPID signature is implemented in Python (Rossum, 1995) and R (R Development Core Team, 2008) and the server is implemented in HTML5, CSS3 and JavaScript.
References:
Baltz et al. The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol Cell. 2012; 46(5):674-90
Castello et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell. 2012; 149(6):1393-406
Kwon et al. The RNA-binding protein repertoire of embryonic stem cells. Nat Struct Mol Biol. 2013; 20(9):1122-30
Bellucci et al. Predicting protein association with long non-coding RNAs. Nat Methods 2011; 8: 444-445
Klus et al. The cleverSuite approach for protein characterization: predictions of structural properties, solubility, chaperone requirements and RNA-binding abilities. Bioinformatics. 2014; 29(22):2928-2930
Li et al. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006; 22(13):1658-9
Pedregosa et al. Scikit-learn: Machine Learning in Python. Journal of Machine Learning Res. 2011; 12: 2825-2830
Nagarajan et al. Prediction of RNA Binding Residues: An Extensive Analysis Based on Structure and Function to Select the Best Predictor. PLoS ONE. 2014; 9(3)