
catRAPID signature Tutorial
catRAPID signature calculates the overall RNA-binding propensity followed by prediction of RNA-binding regions.
Submission
The server automatically assigns a reference number to each submission. It takes as input a protein sequence in FASTA format that can be pasted in the dedicated form. The user can provide an email address (optional) to receive a notification when the job is completed. In addition, the form can be pre-populated by clicking on the provided sample data at the bottom of the submission page.
General information
The algorithm has been trained on proteins with a sequence length between 60 and 2645 amino acids.
Due to technical reasons the algorithm can handle longer than 60 amino acids. Processing a single protein takes 1-3 minutes on average, depending on sequence length.
When multiple sequences are submitted, 10 proteins require around 15-20 minutes, where variations might be caused by number of concomitant submissions.
We restrict the use of the server to 100 proteins, to guarantee that each run will be finished within 3 hours.
There is no restriction with respect to the maximum sequence length, although the total number of submitted characters is restricted to 100000.
Browser compatibility
We ensure that our algorithm works on latest versions of Mozilla Firefox and Google Chrome. We are relying on the latest web technologies and we do not require any extra plugin installation.
Should you experience any problems when uploading files, please ensure that your internet connection works properly and there are no proxies/firewalls blocking the transfer in between you and our servers.
Multiple protein submission
It is also possible to submit more than one protein sequence at a time. In this case, the email address is REQUIRED. The sequences (in FASTA format) can be pasted in the dedicated form and the results will be send to the user via email.
Output and interpretation
After the submission is completed, the algorithm processes the protein sequence in two steps:
i) analysis whether a protein has RNA-binding abilities or not
ii) characterization of putative RNA-binding regions
After the profile calculation, a binary classifier (Pedregosa et al., 2011) assigns an overall score to the sequence, indicating whether the submitted protein is RNA-binding or not. The overall prediction score is shown at the beginning of the page and ranges from zero to one. If the submitted sequence produces an overall score lower than 0.5, indicating no RNA-binding activity, the server stops with the analysis. The example below refers to a non RNA-binding protein (Castello et al.; 2012).

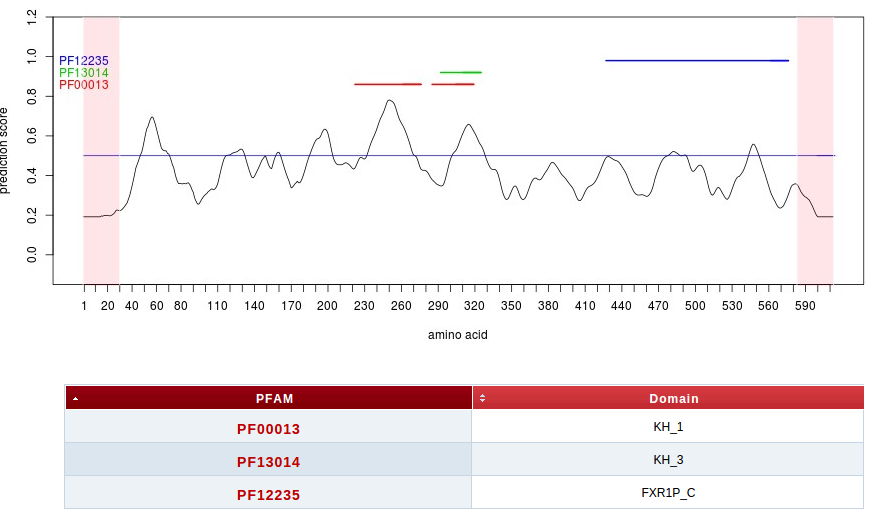
If the submitted sequence obtains an overall score above 0.5, the protein is interpreted as a potential RNA-binding protein and the server continues with the detection of putative RNA-binding regions. The algorithm identifies RNA-binding regions using similarity of physico-chemical profiles with PFAM domains. For this purpose a machine learning method, trained on binding and non-binding sites, calculates for each amino acid the propensity to be part of a binding region or not. Finally the algorithm produces a plot, showing the amino acid sequence on the x-axis and the propensity score on the y-axis.
A threshold (blue line) of 0.5 distinguishes between hot spots and uninteresting zones along the protein sequence.
Annotated binding regions, when present, are also highlighted (using HMMER). The detected PFAM identifiers are listed in a small table below the plot and a link to the respective PFAM summary is provided.
As shown in the following example of Fragile X Mental Retardation Protein (FMRP) our method correctly identifies RDs (two central KH domains and RGG box at 527-552 amino acids) as well as a novel RD at the N-terminus. Importantly the N-terminus interacts with RNA (Myrick et al., 2014) but it is not annotated in PFAM. Also, the RGG box is known to have an accessory role in RNA binding (Bagni et al, 2005) and shows lower propensity scores.
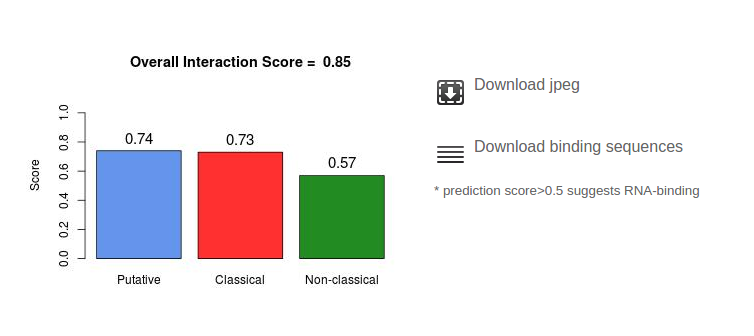
In addition to the overall score, three classifiers are used to assign specific propensities to the Classical, Non-classical and Putative categories. The classifiers have been trained on individual datasets. The division in RNA-binding categories is provided in the Supplementary of Kwon et al., 2013. The scores are visualized in a bar-plot at the bottom of the page. The scores range from zero to one and reflect the propensity to be associated with one of the classes. The user can download both, the plot of the detected binding regions and/or the respective amino acid sequences in FASTA format.
The user has also the opportunity to analyse the predicted interactome of the binding regions. A direct link to catRAPID omics tool (Agostini et al., 2013) is provided and the detected binding regions are automatically uploaded for calculation. Binding regions shorter than catRAPID minimal requirment (i.e., 50 amino acids) will be expanded to 50 amino acids.

Multiple protein submission output
The output of the multiple protein submission is a text file mailed to the user.
The file contains the interaction score for all the submitted proteins and the detected binding regions.

References:
Baltz et al. The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol Cell. 2012; 46(5):674-90
Castello et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell. 2012; 149(6):1393-406
Kwon et al. The RNA-binding protein repertoire of embryonic stem cells. Nat Struct Mol Biol. 2013; 20(9):1122-30
Boratyn et al. BLAST: a more efficient report with usability improvements. Nucleic Acids Res. 2013; 41(Web Server issue):W29-33
Finn et al. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011; 39(Web Server issue):W29-37
Bellucci et al. Predicting protein association with long non-coding RNAs. Nat Methods 2011; 8: 444-445
Klus et al. The cleverSuite approach for protein characterization: predictions of structural properties, solubility, chaperone requirements and RNA-binding abilities. Bioinformatics. 2014; 29(22):2928-2930
Pedregosa et al. Scikit-learn: Machine Learning in Python. Journal of Machine Learning Res. 2011; 12: 2825-2830
Bagni et al. From mRNP trafficking to spine dysmorphogenesis: the roots of fragile X syndrome. Nature Reviews Neuroscience 2005; 6:376-387
Myrick et al. Human FMRP contains an integral tandem Agenet (Tudor) and KH motif in the amino terminal domain. Hum Mol Genet. 2015
Agostini et al. catRAPID omics: a web server for large-scale prediction of protein-RNA interactions. Bioinformatics. 2013 Nov 15;29(22):2928-2930