CROSS Tutorial
The CROSS (computational recognition of secondary structure) algorithm predicts the secondary structure propensity profile of an RNA sequence at single-nucleotide resolution.
Submission
The server automatically assigns a reference number to each submission. As input the user can paste one or multiple RNA sequences in FASTA format into the dedicated form. The user can provide an email address (optional) to receive a notification when the job is completed. The algorithm will process only standard nucleotides (ACGTU). Do not use symbols like !@#$% etc.

The user can choose between five different models, each trained on a dataset from one specific organism and technique. The datasets are described in the documentation section. Each selection allows the user to specify the model to predict the RNA secondary structure (RSS) propensity based on the specific characteristics of a technique or an organism: PARS on yeast (Kertesz et al. 2010), PARS on human (Wan et al., 2014), icSHAPE on mouse (Spitale et al., 2015), SHAPE on HIV (Watts et al., 2009). Due to the high quality of structural experiments, we added a model trained on NMR/X-ray data (Andronescu et al., 2008). The Global Score model is a consensus of these 5 models (see Documentation).
As any technique produces different RSS measures (NMR identifies the interacting pairs, PARS is based on V1 and S1 enzymatic activity, SHAPE requires different chemical-probes), this selection may depend on the experimental aim of the analysis.
In the CROSS homepage, the 5 models are ranked by cross-validation accuracies (see Documentation).
We suggest the user to use the Global Score model since it exploits all the methods and is thus not biased by single experimental specificities. In addition to the secondary structure propensity profile, we also provide the secondary structure computed by RNAstructure (Reuter et al., 2010) using CROSS predictions as soft constraints for sequences with less than 500 nucleotides.
Alternatively, since SHAPE is becoming a widely used technique and the icSHAPE-mouse model was trained on a large amount of cases, we also recommend the user to choose this model.
See the Documentation for more details about the datasets.

The form can also be pre-populated by clicking on the provided sample data at the bottom of the submission page. The algorithm will compute the complete secondary structure propensity profile of Xist in less than 1 minute using Global Score.

General information
The algorithm has been trained on RNA sequences of different lengths and characteristics. Due to technical reasons the algorithm can only handle sequences longer than 13 nucleotides (we set the minimum to 25 nucleotides to generate a reasonable profile, see Documentation). Processing a single RNA of 200 nucleotides takes less than 1 minute. As for the time required to predict RSS propensities of multiple sequences, processing 10 sequences (200 nt each) required around 1 minute (test_10), and a set of 100 sequences can be processed in 2 minutes (test_100). To avoid overloading the server and to generate the output file in a convenient format, the webserver is optimized to process a maximum of 300 sequences with a total of 60'000 characters (3 minutes).
There is no restriction with respect to the maximum sequence length, but we suggest submitting less than 1'000 sequences at once.
Browser compatibility
We ensure that our algorithm is compatible with the latest versions of Mozilla Firefox and Google Chrome. We are relying on the latest web technologies and we do not require any extra plugin installation. Should you experience any problems when uploading files, please ensure that your internet connection works properly and there are no proxies/firewalls blocking the transfer in between you and our servers.
Multiple sequences submission
You can also paste multiple sequences (in FASTA format) into the dedicated form. The output will be stored in a .zip file that the user can easily download with one click.
Output and interpretation
After the submission is completed, the algorithm processes the RNA sequences using the experimental model previously selected. For each sequence of length n the algorithm computes its secondary structure propensity profile at single nucleotide resolution, from nucleotide 7 to n-7. To calculate the scores for the first and last 6 nucleotides we employ periodic boundary conditions: we append a copy of the last 6 nucleotides at the 3’ end to the 5’ end and vice versa. The scores of the first and last 6 nucleotides are then calculated using fragments containing nucleotides from the respective other end. The predicted secondary structure propensity scores lie in the range [-1, 1]. A score in the range (0, 1] means that the nucleotide is predicted to be in a double-strand, while a score in the range [-1, 0) indicates that the nucleotide is single stranded. To generate a better visualization of the profile, we apply a length-dependent dynamic smoothing. A scrolling table shows a sample of the total nucleotides (depending on the smoothing window) with both the raw and the smoothed propensity score. The user can easily download the table with the complete data (raw and smoothed) by clicking the provided link. The symbol “-” indicates nucleotides outside of the smoothing window where the smoothed score is not available. Please refer to the raw propensity score for these cases.

To show the distribution of the RSS propensity scores, the algorithm generates a visualization of the profile using R. The profile is smoothened using a running mean with a dynamic window size linearly dependent on the length of the sequence. This image can be downloaded in PNG format.
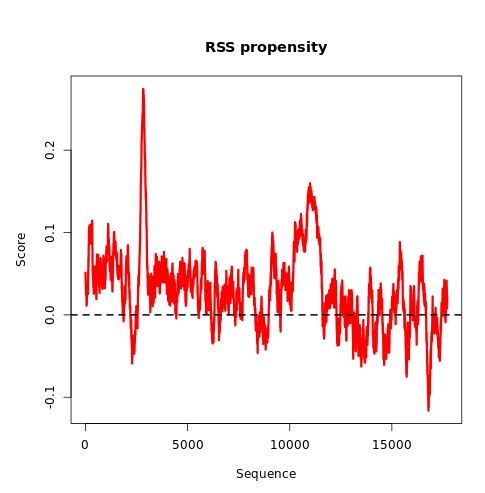
The algorithm generates a simple visualization of RNA secondary structure profile in a scrolling window. The symbol "." means that the nucleotide is probably single-stranded, while the symbol "|" indicates a higher propensity to be double-stranded.

Multiple sequences submission output
The user can also paste multiple RNA sequences in the form, however the output page will be slightly different. The webpage will show the link to download the output of the submission as a zip file (Submission.zip).

After unzipping the file, you can find the visualizations of the profiles as sequence_name.png in Submission/Graphs and the profile tables as sequence_name.txt in Submissions/Profiles.
Global Score
Global Score is a consensus model that we integrated in RNAstructure (Reuter et al., 2010). The profile visualization is generated in the same way as for the other models. The structure is generated using RNAstructure with Global Score as constraints, thus it will be represented using the "dots and brackets" notation.
The profile predicted by CROSS and the structure computed by RNAstructure could be different since RNAstructure uses Global Score as a soft constraint.

In case of multiple submissions, the structures predicted by RNAstructure are saved in .ct files inside the folder Submission/Structures.
Note that due to the complexity of the configurational space, only sequences shorter than 500 nucleotides will be processed using RNAstructure (the time to run Global Score can be 10 times slower). The structure of sequences longer than 500 nucleotides will be displayed in the standard way instead of as “dot and brackets”.
References:
Kertesz et al. Genome-wide measurement of RNA secondary structure in yeast. Nature. 2010 Sep 2;467(7311):103-7. doi: 10.1038/nature09322
Wan et al. Landscape and variation of RNA secondary structure across the human transcriptome. Nature. 2014 Jan 30;505(7485):706-9. doi:10.1038/nature12946
Andronescu et al. RNA STRAND: the RNA secondary structure and statistical analysis database. BMC Bioinformatics. 2008 Aug 13;9:340. doi: 10.1186/1471-2105-9-340
Watts et al. Architecture and secondary structure of an entire HIV-1 RNA genome. Nature. 2009 Aug 6;460(7256):711-6. doi: 10.1038/nature08237
Spitale et al. Structural imprints in vivo decode RNA regulatory mechanisms. Nature. 2015 Mar 26;519(7544):486-90. doi:10.1038/nature14263
Reuter et al.,. RNAstructure: software for RNA secondary structure prediction and analysis. BMC bioinformatics. 2010. doi:10.1186/1471-2105-11-129