- Introduction
- Selection of in vivo data
- Cross-validation of the models
- Predicting the RNA structure in vivo using information from the interaction with proteins
- Training of the networks
- Xist in vivo structure
-
CROSSalive Documentation
Introduction
Through analysis of transcriptome-wide data, we built a method for the prediction of RNA secondary structure in vivo. One key element in our approach is the prediction of protein interactions, which allows us to better mimic the complex cellular environment also using contributions of thermodynamic approaches such as Vienna (Gruber et al. 2008). For the first time we introduce a large-scale analysis of in vivo data and highlight a relationship between proteins and structure, predicting in vivo data with an accuracy of 0.80 or higher.
Selection of in vivo data
Dymetil-Sulfate (DMS) can study the contributions of adenine and cytosine to RNA structure (Mortimer et al. 2014) and the 1M7 and NMIA reagents of Selective 2′ hydroxyl acylation analyzed by primer extension (SHAPE) have poor solubility and reactivity (Lee et al. 2017), icSHAPE is, at present, the most reliable in vivo technique by using the chemical probe NAI-N3 (Spitale et al. 2015). Following the strategy applied on our previous work (CROSS; Delli Ponti et al. 2017), we selected 100'000 RNA regions encoding the highest icSHAPE signal for single- (reactivity=1) and double-stranded conformation (reactivity=0 or lack of signal). These fragments were used to assess the sequences contribution during the training process.
Cross-validation of the sequence-based models
Artificial neural networks (ANN) were initially trained only using sequence contributions: icSHAPE in vitro data and icSHAPE in vivo with and without Mettl3 knock down (m6a+/m6a-) (Spitale et al. 2015). Three ANNs (in vitro, in vivo m6a+, in vivo m6a-) were trained on the same conditions (training and testing sets of the same size), and cross-validated between each other. The in vitro model is the one with the best performances in in 10-fold validation during the training step, showing how it is easier to predict the structure in a controlled environment outside the cell (0.86 accuracy or ACC). However, during the cross-validation with the other datasets, we noticed that the in vitro model is not able to correctly predict in vivo datasets. This is because of the complexity of the in vivo conditions, which cannot be predicted using sequence information only (Delli Ponti et al. 2017), since RNA structure is altered by the presence of proteins. To correctly predict the RNA secondary structure in vivo, we decided to integrate the protein contributions into the algorithm.
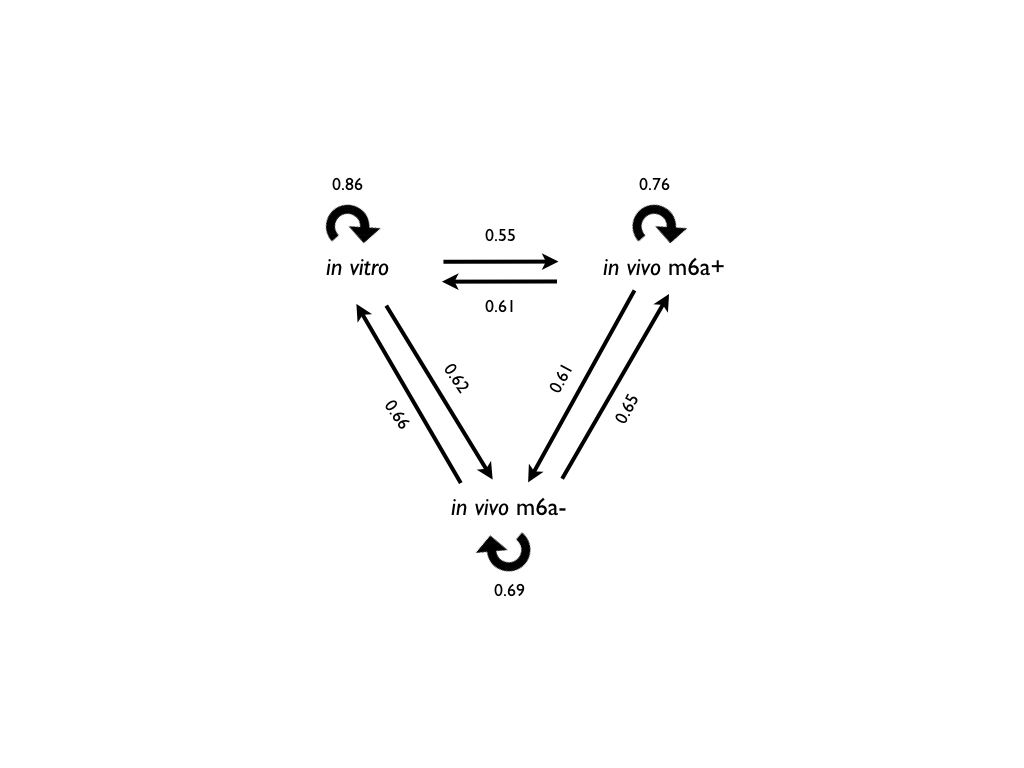
Predicting the RNA structure in vivo using information from the interaction with proteins
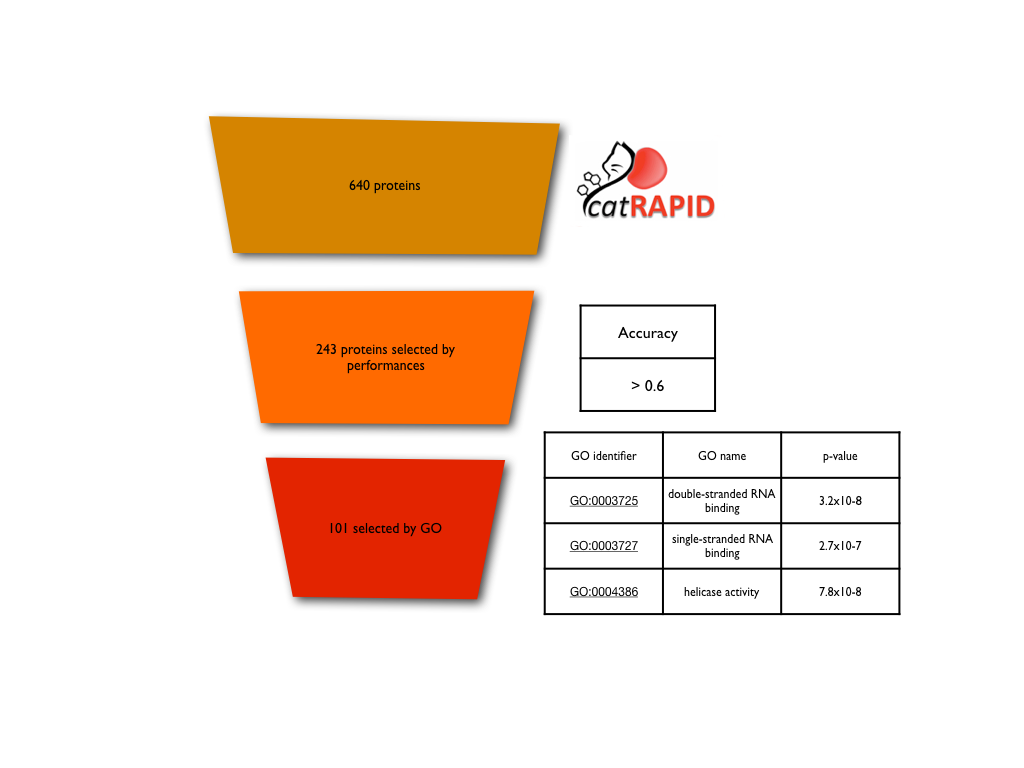
We used catRAPID to predict the interaction of several regions from 640 RNA-binding proteins (Agostini et al. 2013) with 200´000 double/single-stranded in vivo RNA fragments (a total of 128 millions of interactions). The protein regions were firstly selected for their ability to discriminate single and double-stranded RNA using icSHAPE data (Accuracy > 0.6). We found that the dataset was enriched for proteins with GO related to RNA structure and further selected cases with strong signals. The same procedure was used for the RNA structure in vivo without m6a modifications (m6a-).
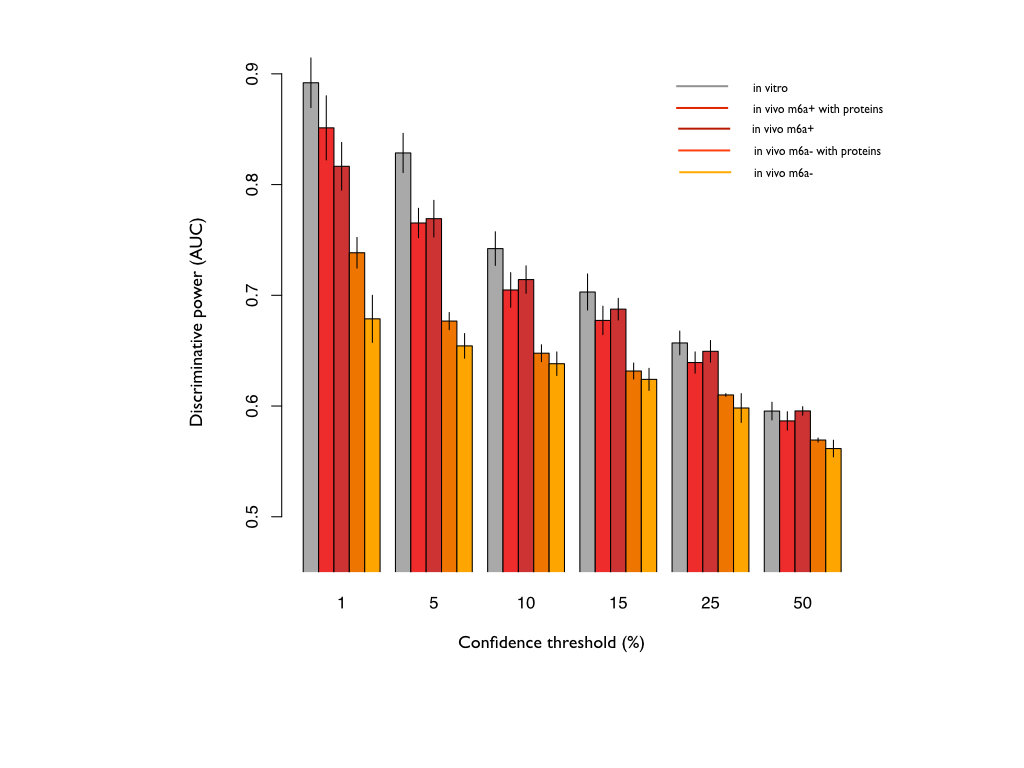
 The selected proteins were included into the training of the algorithm, to complement the information coming from the sequences. The scores show a monotonic trend with stronger propensities (e.g., top 1% of single-stranded and top 1% of double-stranded regions) discriminating better than lower propensities (e.g., top 5% of single-stranded and top 5% of double-stranded regions). Note that 50% corresponds to the median.
The selected proteins were included into the training of the algorithm, to complement the information coming from the sequences. The scores show a monotonic trend with stronger propensities (e.g., top 1% of single-stranded and top 1% of double-stranded regions) discriminating better than lower propensities (e.g., top 5% of single-stranded and top 5% of double-stranded regions). Note that 50% corresponds to the median.
Using also the protein contributions leads to an improvement of the predictive power of the algorithm, up to an ACC of 0.88 during the 10-fold validation), with even a more significant improvement in m6a-.
Training of the networks
For more information about the component of the ANN, check our previous paper (CROSS; Delli Ponti et al. 2017). We selected the 100’000 fragments of 51 nucleotides with the middle nucleotides with a higher propensity to be single-stranded for icSHAPE reactivity, and the 100’000 with a higher propensity to be double-stranded. The window size is enough to capture the combinatorial complexity of icSHAPE data on mouse transcriptome (i.e., 451 > 1.2x107), which is also an accepted size for catRAPID algorithm. The sequence component was coded using a ‘one hot encoding’ procedure, where each nucleotide is converted in a 4mer notation: A = (1, 0, 0, 0), C = (0, 1, 0, 0), G = (0, 0, 1, 0) and U = (0, 0, 0, 1). This approach was used to train the ANN using the sequence only. To study the protein binding, the catRAPID score of the 101 most discriminative proteins was integrated in the training step. First, the youden cut-off was computed for each protein on the complete dataset of RNA fragments (single/double-stranded). Then the score of each protein was normalized using the cut-off, setting the scores higher to 1, and the lower to -1. The following 101 normalized score were integrated for each RNA with the information coming from the sequence, for a total training complexity of 305 variables. Similarly, the in vivo-m6a was trained using 81 discriminative proteins for a total of 285 input variables. The networks were trained using a 10-cross fold validation, keeping always balanced the positive and negative set. Redundancy of equal sequences was removed both intra- and inter-sets.
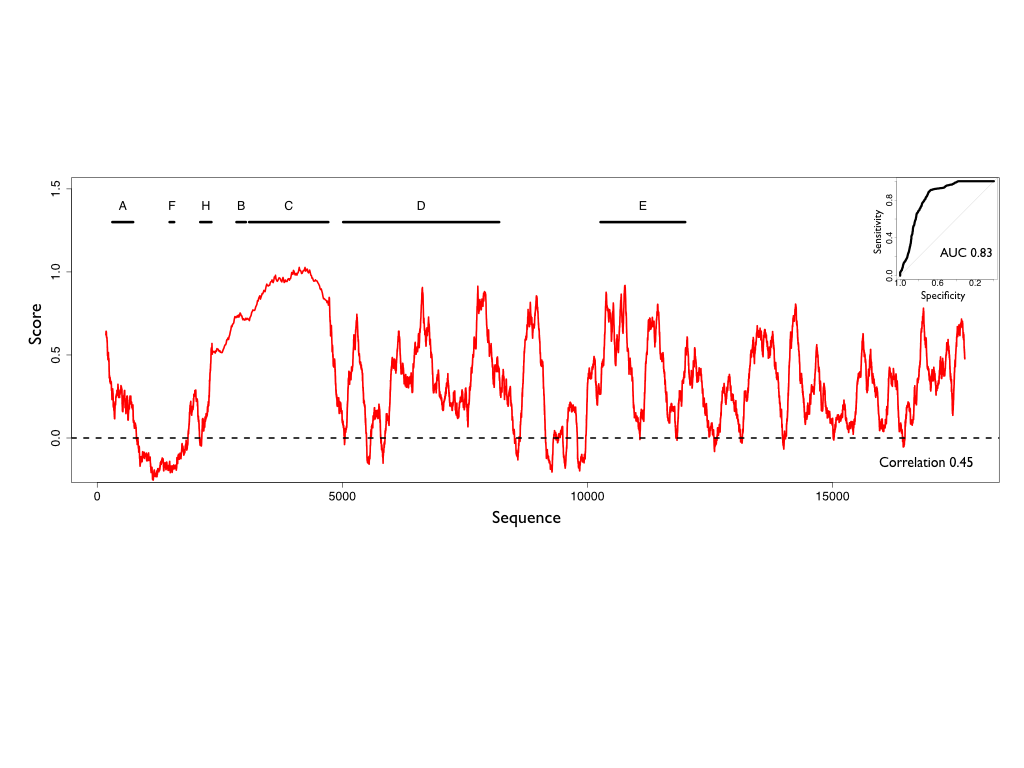
Xist in vivo structure
CROSSalive was tested against a complete independent in vivo SHAPE-Map experiment on murine Xist (~17900 nt; not present in icSHAPE experiments; Smola et al., 2016). The in vivo m6a- is the most reliable model to use, since lncRNAs are usually less methylated than mRNAs. Mettl3 is less abundant in trophoblasts (cell line used in SHAPE-Map), which indicates Mettl3-mediated m6a modifications are less relevant. CROSSalive profile has a correlation of 0.45 with SHAPE-Map data on Xist. The algorithm achieves an AUC of 0.83 on the 30% highest-confidence single and double-stranded regions ranked by SHAPE reactivities. The m6a- model trained only on sequence data achieves only an AUC of 0.53 on Xist, showing how the contribution of proteins improves the predictive power in vivo.