Introduction
ccSOL omics allows fast and accurate large-scale predictions of protein solubility. The algorithm exploits a list of physico-chemical scales, such as hydrophobicity/hydrophilicity, coil/turn/disorder and alpha-helix to compute propensity profiles for each protein.
The profiles are used to compute the overall solubility propensity by means of an algorithm whose architecture is explained in Tartaglia et al., 2009 .
.
In this tutorial you will find information on:
ccSOL omics Tutorial
Submission form

As soon as the ccSOL omics module is selected, the server generates automatically a unique reference number for the submission. The user can optionally supply a custom submission label and an email address to receive the notification of job completion:
Please note that the email address is optional and that the Tor browser can be used to run prediction in complete anonymity.
The user is asked to provide a list of protein sequences in FASTA format as input file. Two options are possible:
A) Text box upload
B) File upload (plain text format)

Once the job has been submitted, the link to the result page will be provided.
The user can pre-populate the form by using two illustrative cases:
A) Human prion (text box upload)

B) Escherichia coli proteins (file upload)

The ccSOL webserver requires the most recent version of the following browser with JavaScript enabled: Chrome, Firefox and Safari. Internet Explorer is not fully supported. If your browser connects through a proxy, please, be aware that you might experience a slow upload of the example data in the query forms.
Examples of output
By clicking on Human Prion link, the Major prion protein (PRIO) proteins will be loaded as FASTA in the textbox.
In this case, where the number of entries is below 500, the output will provide the table of the solubility scores along with the graphical representation of the result (PDF files) in the Distributions column:

The solubility scores provided in the table are computed using Fourier transform on solubility profiles and a neural network.
Although the algorithm has been train on a huge number of sequences, it is possible that a bias can arise in the prediction of proteins with sequence sizes outside the training set range. To take into account of this factor and provide more reliably predictions, each prediction is associated with a reliability score, represented by 1 to 3 stars:
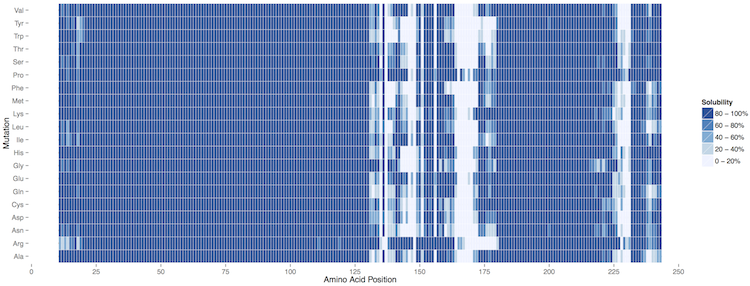
of the distribution;Additional plots are provided for the SVM classification: Profiles of protein solubility and maximal/average susceptibility are available for download (PDF files) in the Profiles column. Protein solubility profiles are generated by computing the solubility of a sliding window of 21 amino acids. In addition, the central position of every window is mutated into all the other amino acids and the maximum (most soluble and least soluble substitutions) and average solubility variations are reported along the sequence.
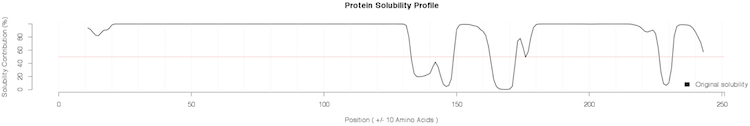
Our algorithm identify the human prion as non-soluble protein in E. coli and correctly spots the fragment 130-170 as the most insoluble within the C-terminus of human PrPtogether with region 231-253 (not present in the mature form). This finding is very well in agreement with what has been discussed
in previous reports (Tartaglia et al., 2005 and Tartaglia et al., 2008)
and with the fact that expression of mammalian proteins in E. coli remains a difficult task and often results in inactive aggregates (Abskharon et al., 2012 and Baneyx et al., 2004).
Indeed, recombinant PrP is poorly soluble and accumulates in inclusion bodies (Mehlhorn et al., 1996).
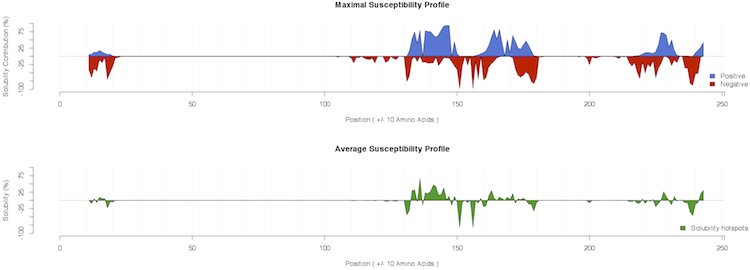
A comprehensive view of all the mutations introduced in the generation of the profiles is available as heatmap (PDF file) in the Susceptibility and as table (txt file) in the Raw Data columns.
As shown in the example, ccSOL omics is able to correctly identify the contributions of single point mutations in the 130-180 region implicated in PrPSc conversion (Corsaro et al., 2012):
some of them decrease the solubility propensity of the protein (e.g. G131V, S132I, R148H, V176I, D178N) (Chakrabarti et al., 2010, Pastore et al., 2005,
Swietnicki et al., 1998 and Hafner-Bratkovič et al., 2011), while others have little or no effect (e.g. P102L, E200K, D202N)
(Swietnicki et al., 1998 and Corsaro et al., 2012).
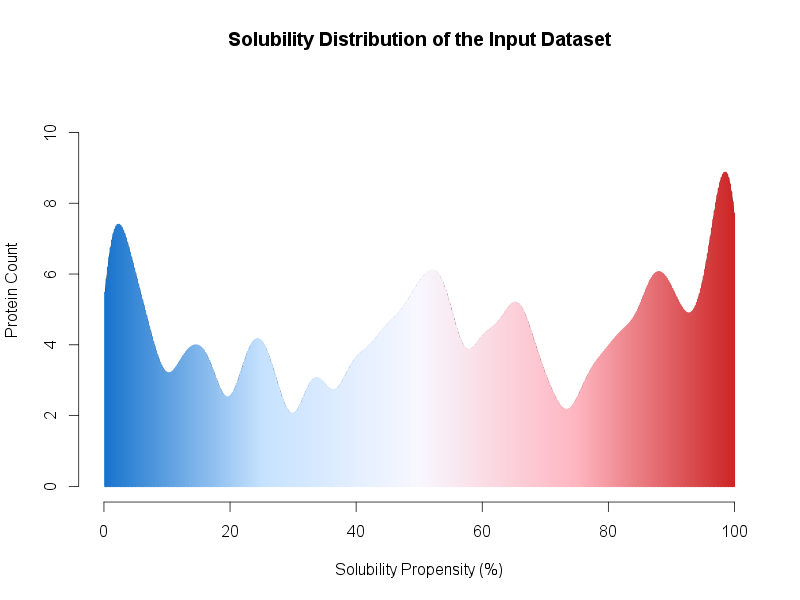
In addition, we provide an example in the file upload submission form. By clicking on the Escherichia coli proteins link, 500 (250 soluble and 250 insoluble) proteins will be loaded as FASTA sequences. Since the total amount of entries is >= 500 entries, once the prediction is completed, the result page will present a distribution of the global dataset solubility (see Figure below) along with a table of the individual solubility propensity (%) score.
Performances
The training has been carried out using a non-redundant dataset (CD-HIT similarity < 30%) of soluble (18'495 entries) and insoluble (18'495 entries) proteins retrieved from Target Track:
- Protein constructs that achieved the soluble status or subsequent stages (including native_diffraction-data, NMR_assigned, phasing_diffraction-data, diffraction, in_BMRB, NMR_structure, crystal_structure, diffraction-quality_crystals, in PDB, crystallized, HSQC) were considered soluble;
- We considered as insoluble all the proteins that did not reach the soluble status and are classified as work stopped.
We found mismatches between Target Track annotations and what is reported in SOLpro and PROSO II datasets.
Differences are due to i) use of older databases, such as pepcDB (now integrated in Target Track); ii) less stringent criteria to define the insoluble status (proteins without "soluble status" before a certain date were considered insoluble).
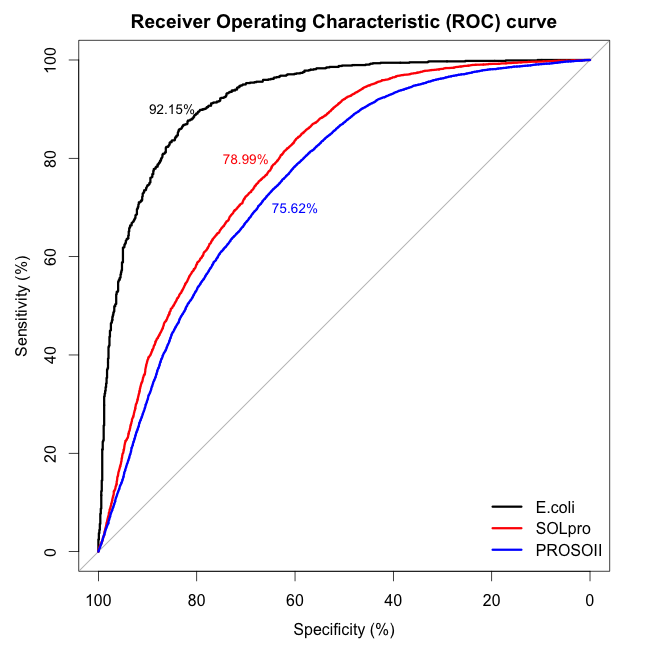
We benchmarked ccSOL omics on three independent datasets [total of 31’760 entries with 30% sequence redundancy]: E. coli (Niwa et al., 2009), SOLpro (Magnan et al., 2009)
and PROSO II (Smialowski et al., 2012)].
The overall accuracy in discriminating between soluble and insoluble proteins is 78% (SOLpro and PROSO II sets contained mismatches with respect to Target Track and were re-annotated).
Using ccSOL omics we investigated the E. coli DnaK/GroEL substrates (chaperone-dependent; Kerner et al., 2005),
amyloid proteins (aggregating; Pawlicki et al., 2008), heterologously/endogenously expressed proteins used in folding
kinetics studies (folding; Tartaglia G.G. and Vendruscolo M., 2010), and independently-folding E. coli proteins (chaperone-independent;
Kerner et al., 2005):
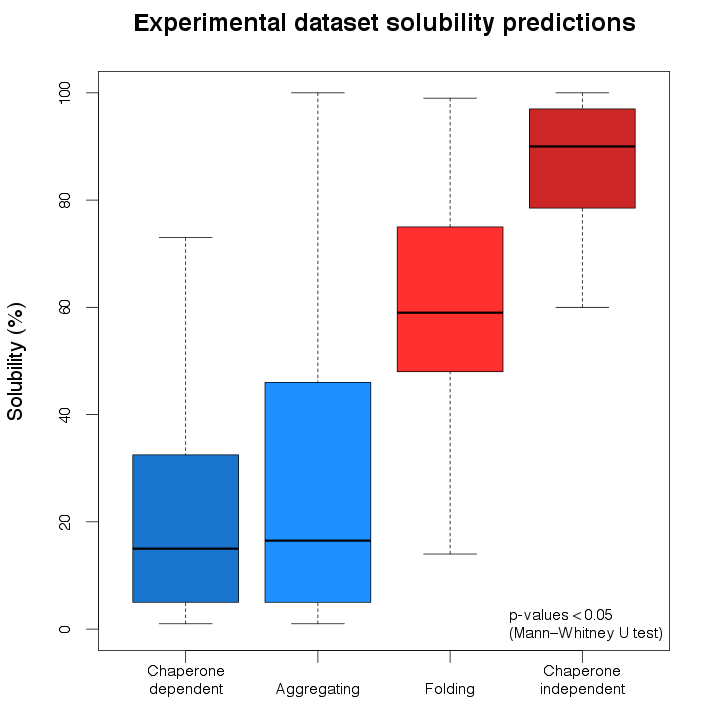
In agreement with the existing evidence, chaperone-dependent and aggregating proteins are predicted as mostly insoluble, while folding and chaperone-independent proteins are classified mainly as soluble.
Train and test data
To reduce the redundancy of the datasets, we filtered each set using CD-HIT at 30% of similarity threshold:
Additional experimental datasets used for testing the performance of the algorithm: