SeAMotE Tutorial
SeAMotE (Sequence Analysis of Motifs Enrichment) allows fast and accurate large-scale de novo motif discovery in nucleic acid sequences.
Submission form
As soon as the SeAMotE module is selected, the server generates automatically a unique reference number for the submission. The user can optionally supply a custom submission label and an email address to receive the notification of job completion:
The user is asked to provide a list of sequences in FASTA format as input file (Positive dataset).
The algorithm exploits a series of pre-generated seed motifs (e.g. ACY = AC[CT]) that are employed in the computation of dataset enrichment and discrimination. In order to focus on specific comparisons, several options are offered to the user.
The first option is the Reference set that will be used for the calculation of motifs enrichment. The Shuffle option will generate a Reference set of same dimension and sequence composition as the Positive set. The Random option will generate a Reference set of same dimension as the Positive set but with random composition (human transcriptome-derived letter frequencies).

The Custom option will exploit a user-provided Reference dataset.
The second option is the Set Coverage that will be used for the calculation of the motif enrichment. The Default option will execute SeAMotE algorithm with a default coverage threshold of 0.75 (i.e., searched patterns covering less then 75% of the dataset will not be extended). Other options will execute SeAMotE algorithm with the coverage threshold chosen by the user.

Finally, the user has to specify if the analysis should be performed only in the given strand (e.g. search for splicing factor binding sites) or if the opposite strand should be checked (e.g. search of transcription factor binding sites) for possible patterns.

Once the job has been submitted, the link to the result page will be provided.
In addition, the user can pre-populate the form by using an illustrative case:
FUS protein recognition motifs from PAR-CLIP data (Hoell, J. I. et al. 2011).
Interpreting the output
Let’s try SeAMotE with one example. Click on the link PAR-CLIP bound/unbound transcripts (FUS protein)
and FASTA sequences of FUS PAR-CLIP bound and not bound RNAs (Hoell, J. I. et al. 2011) will be uploaded as Positive and Reference set respectively.
RNA-binding protein FUS (Fused in Sarcoma) is a component of nuclear riboprotein complexes and the pathological term for frontotemporal dementia (FTD) syndrome.
SeAMotE is able to identify significantly discriminated recognition motifs of FUS (best motif p-value: 2.12x10-309; Discrimination: 61%).
Once the prediction is completed, the result page will present a summary of the job:
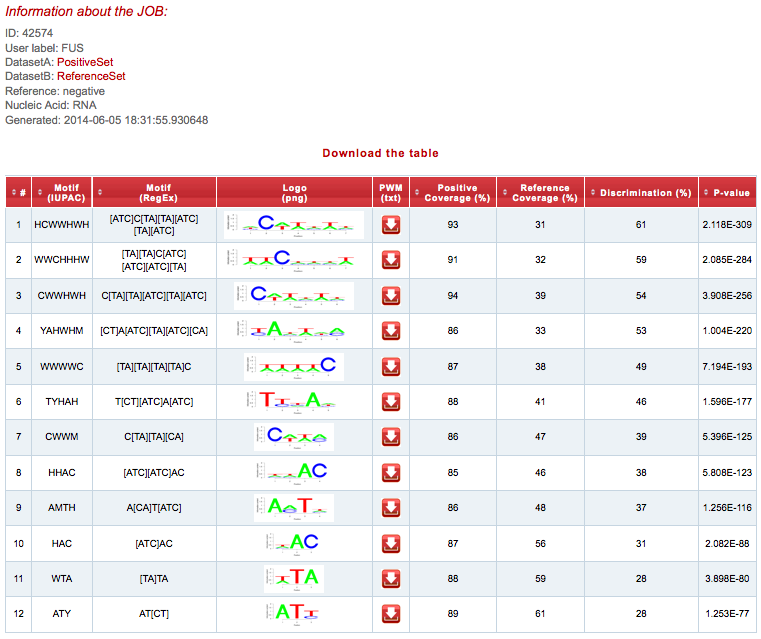
The output consists of a table listing the most significantly discriminated motifs in the Positive set. The fist and second columns describe the motif in IUPAC nucleotide code and regular expression (regex) format, respectively.
| IUPAC nucleotide code | Base |
|---|---|
| A | Adenine |
| C | Cytosine |
| G | Guanine |
| T (or U) | Thymine (or Uracil) |
| R | A or G |
| Y | C or T |
| S | G or C |
| W | A or T |
| K | G or T |
| M | A or C |
| B | C or G or T |
| D | A or G or T |
| H | A or C or T |
| V | A or C or G |
| N | any base |
The third and the fourth columns show the logo representation and the positional weighted matrix (PWM) download button, respectively. The logo can be obtained as a PNG image and the PWM as a TXT file. The fifth and sixth columns describe the motif coverage (i.e., motif containing sequences:total set sequences ratio) in the Positive (Sensitivity) and Reference (1-Specificity) sets respectively. The seventh column describes the percentage of discrimination (i.e., the Youden's Index = Sensitivity + Specificity - 1). The eighth column describes the p-value (Fisher's exact test) associated with the motif.
Using the link above the table, the user can download the complete table in TXT format.
Performances
We benchmarked the algorithm on in vivo CLIP-seq data assembled from literature (see references below). Here, we report the performances with respect to DREME (Bailey TL., Bioinformatics., 2011), a very well established tool for discriminative motif discovery. SeAMotE outperforms DREME in discovering motifs with both higher discrimination and higher statistical significance.
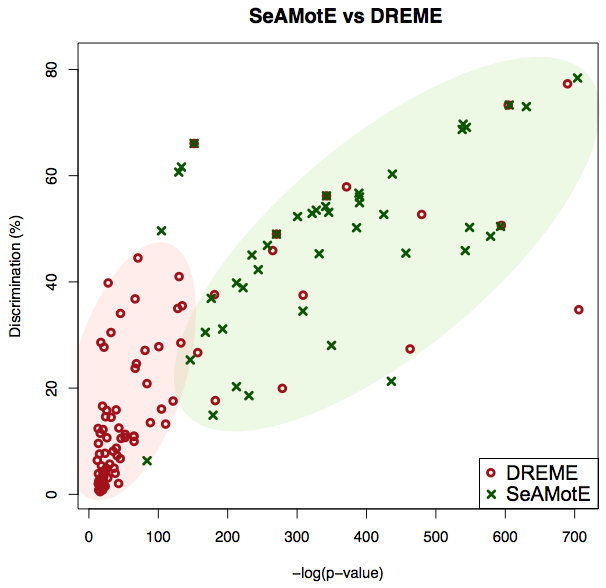
The illustrative example of SeAMotE and DREME performances on the FUS dataset (Hoell, JI. et al. 2011) is provided.
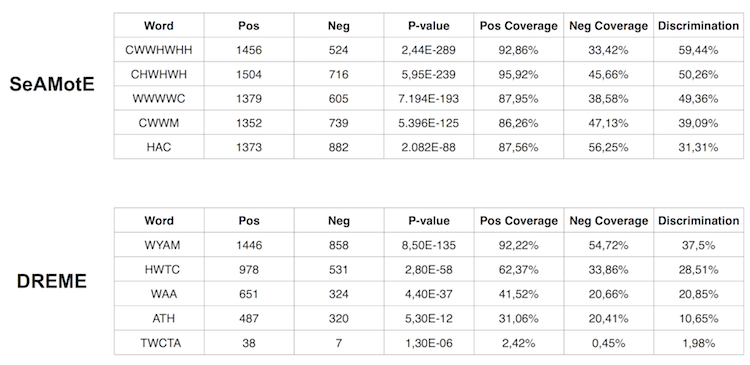
SeAMotE and DREME performances on PAR-CLIP datasets
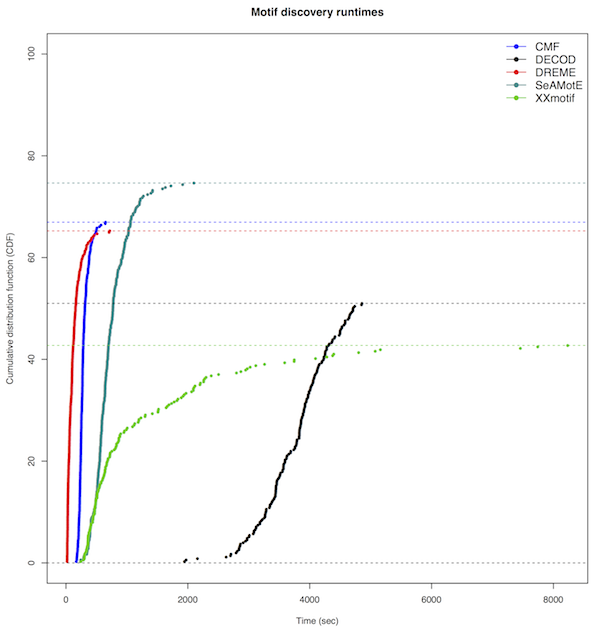
Motif discovery time performance
Motif discovery runtimes of CMF (Mason, MJ. et al., Bioinformatics., 2010), DECOD (Huggins P. et al., Bioinformatics., 2011), DREME (Bailey TL., Bioinformatics., 2011), XXmotif (Luehr S., Nucleic Acids Res., 2012) and SeAMotE algorithms are plotted for 351 transcription factor (TF) ChIP-seq data sets (Harrow J. et al., Genome Res., 2012). The cumulative distribution function represents the percentage of Jaspar (Mathelier, A., et al., Nucleic Acids Res., 2014) and Jolma, A. et al., Cell, (2013) annotated TF motifs that are recovered using TOMTOM (Tanaka, e. et al., BMC bioinformatics., 2007).