CROSSalign Tutorial
CROSSalign predicts the structural similarities of two RNA profiles of different length. The tool is based on CROSS and a Dynamic Time Warping algorithm (see Documentation).
Submission
The server automatically assigns a reference number to each submission. As input the user can paste one or two RNA sequences in FASTA format into the dedicated form. Please keep in mind that the RNA sequence in the first form will be used as input for the OBE-DTW and the Fragmentation modes. When using the OBE-DTW the first sequence should be the shorter. The user can provide an email address (optional) to receive a notification when the job is completed. The algorithm will process only standard nucleotides (ACGTU). Do not use symbols like !@#$% etc.

The algorithm can be launched with 3 different modes, each of them is a specific variation of the DTW algorithm. The “standard-DTW” is recommended to compare profile of very similar length.
OBE-DTW (open begins and ends) is a specific mode to search a smaller profile inside a bigger one. This is the recommended modality when comparing profile of very different sizes. Please keep in mind that the sequence inside the form RNA input 1 will be the one searched inside RNA input 1, so the sequence in RNA input 1 should be smaller than the other. We advice to use OBE-DTW when one of the two sequences is at least > 5 times longer than the other.
The “fragmented OBE-DTW” is a specific modality to search unknown secondary structure domains of one profile inside the other. The secondary structure RNA input 1 sequence will be fragmented with a non-overlapping window of 200 nucleotides. Each of the 200 nucleotides profiles will be then searched against the other sequence. This is the recommended mode when the user is not interested in the global similarity between the secondary structure profiles, but wants to search an unknown domain conserved in the two molecules.
A minimum length of 600 nucleotides it is recommended for fragmentation.

The "dataset" mode allows the user to search a single sequence inside all the lincRNAs of a specific organism. The shorter profile of each couple will be searched in the bigger one following the OBE-DTW procedure.
The organisms available are Human, Mouse, Rat, Macaque and Zebrafish. The lincRNAs where downloaded using Biomart (Ensemble 82). New organisms and updated versions will be regularly added.

For more details please check the Documentation.
The form can also be pre-populated by clicking on the provided sample data at the bottom of the submission page.

General information
The algorithm has been trained on RNA sequences of different lengths and characteristics. Due to technical reasons the algorithm can only handle sequences longer than 13 nucleotides (we set the minimum to 25 nucleotides to generate a reasonable profile, see Documentation). Processing two RNAs of 500 nucleotides takes less than 1 minute with standard DTW. The fragmentation of a big RNA (9000nt) against a big profile (9000nt) should take no more than 12 minutes. The dataset mode can take longer, for a sequence of 400nt: 10 minutes against all the lincRNAs of Zebrafish, 1 hour against Mouse and up to 3 hours for Human.
There is no restriction with respect to the maximum sequence length.
Browser compatibility
We ensure that our algorithm is compatible with the latest versions of Mozilla Firefox and Google Chrome. We are relying on the latest web technologies and we do not require any extra plugin installation. Should you experience any problems when uploading files, please ensure that your internet connection works properly and there are no proxies/firewalls blocking the transfer in between you and our servers.
Multiple sequences submission
The webserver supports only 1 sequence input for each specific form.
Output and interpretation
The output of CROSSalign changes in base of the specific DTW mode that the user selected.
Standard-DTW
The main output is the structural distance, the overall value establishing the similarity between the two input structures. The closer the distance is to 0, the higher the similarity in term of secondary structure, where 0 means identical structural profiles. According to our analysis, RNA molecules with a structural distance of 0.095-0.10 or higher are to be considered different in term of secondary structure.
For the significance of each comparison, see section p-values.

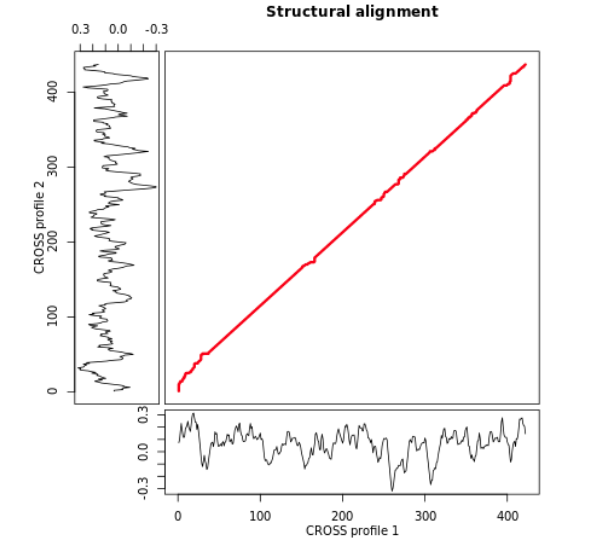
The main plot shows the overall structural similarity of two profiles.
On the two axes the user can see the structural profiles obtained with CROSS Global Score for the two RNAs
(score >0 means a double-stranded nucleotide; <0 single-stranded; see Documentation: CROSS algorithm).
For an easy visualization the profiles of CROSS are smoothed using the same function of CROSS webserver (see CROSS: Tutorial).
The similarity is represented by the red path in the figure, obtained with the index function of the DTW package (see Documentation: DTW).
Vertical or horizontal paths are to be considered gaps, while diagonal paths highlight similar regions of the two profiles.
The matrix used for generating the image can be downloaded at a link provided by the server.
OBE-DTW
The structural distance is also reported in the OBE-DTW mode, as previously explained. Since OBE-DTW allows the identification of the best starting/ending points of a match, the optimal match is reported in term of coordinates on the bigger profile (RNA input 2).
Please keep in mind that the length of RNA input 1 is always smaller than that of RNA input 2 (the server automatically orders the files), and RNA input 1 will therefore searched inside RNA input 2 (see Documentation: DTW).

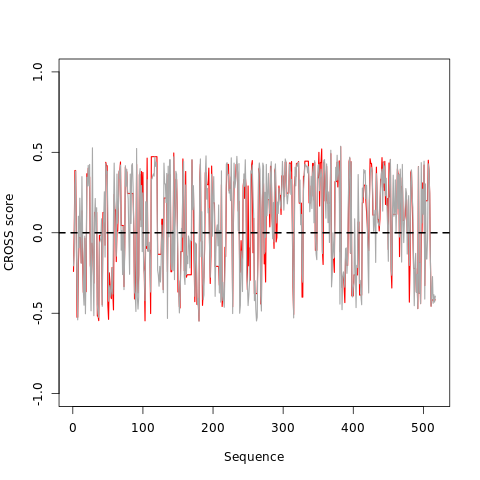
The main plot shows the CROSS profiles of the optimal matching region selected by the OBE-DTW algorithm. To keep the gaps introduced by the algorithm, the two profiles are not smoothed. Big profiles can be difficult to visualize.
Fragmented OBE-DTW
The fragmentation is a particular form of OBE-DTW optimized to search all the possible structural domains of a particular sequence against an entire sequence.
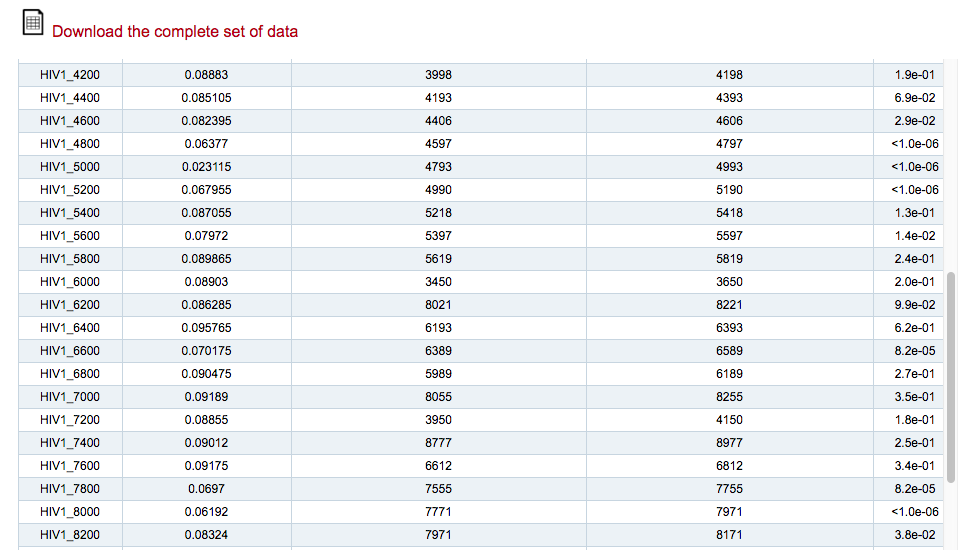
The main output is a scrolling table reporting the structural score, the starting match, the ending match and the p-value for all the fragments of RNA input 1 against RNA input 2. All the values are computed with the same procedures used for OBE-DTW.
The table can also be downloaded in a .txt file.
All the images are generating using using R and can be downloaded in PNG format.
Dataset
The output can be downloaded in a .zip file selecting the specific link. The table has the same format and characteristics of the one obtained using fragmentation. The only difference is that each match is between the input and all the lincRNAs of the specified organism.
Also keep in mind that if the target is shorter than the input sequence, the algorithm will search with an OBE-DTW algorithm the target inside the input profile.
