catRAPID omics v2.1 Tutorial
The catRAPID omics v2 module allows the prediction of RNA-protein partners in a number of model organisms. It is also possible to compute the interaction propensities of a custom set of proteins versus a custom set of RNAs. In the home page, the user is requested what kind of analysis should be performed.
Submission form (protein(s) VS transcriptome)
As soon as the submission form is accessed, the server automatically generates a unique reference number for the submission. The user can optionally choose a custom submission label:

The algorithm exploits a series of pre-calculated reference datasets that could be employed in the computation of the interactions. In order to focus on specific associations, two options are provided to the user:
The first option is the class of RNAs that will be used in the computation.

The second option is the organism of origin for the transcriptome used in the calculation of the interaction propensities. If “circular RNAs” class was selected, choice is restricted to the three species for which precompiled circRNA libraries are available.

The user can provide an email address to receive the notification of job completion (optional, strongly suggested when submitting multiple sequences).

Finally, the user submits up to 10 query protein sequences in FASTA format either by pasting them in the text area (maximum 25000 characters allowed)...
...or by uploading a file.

By clicking on the “Submit” button, the job is submitted. As soon as data is loaded, the user can access to results by clicking on "result".
Submission form (transcript(s) VS RNA-binding proteome)
As soon as the submission form is accessed, the server automatically generates a unique reference number for the submission. The user can optionally choose a custom submission label:
The algorithm exploits a series of precalculated reference datasets that could be employed in the computation of the interactions. In order to focus on specific associations, two options are provided to the user:
The first option is the class of input RNA molecules. Each input RNA will be aligned to the selected precompiled RNA library and assigned the orthology relationships of the most similar RNA, if % Identity is higher than 70%. If “Undefined” is selected, evolutionary conservation analysis will not be performed, otherwise query RNAs will be aligned to the specified set of RNAs to infer orthology relationships. If “circular RNAs” is selected, input RNAs will be fragmented as circular molecules.

The second option is the organism of origin for the RNA-binding proteome used in the calculation of the interaction propensities. If “circular RNAs” class was previously selected, choice is restricted to the three species for which precompiled circRNAs libraries are available.
The user can provide an email address to receive the notification of job completion (optional).
Finally, the user submits up to 10 query RNA sequences in FASTA format either by pasting them in the text area (maximum 25000 characters allowed)…

...or by uploading a file.

By clicking on the “Submit” button, the job is submitted. As soon as data is loaded, the user can access to results by clicking on "result".
Submission form (custom protein set VS custom transcript set)
As soon as the submission form is accessed, the server automatically generates a unique reference number for the submission. The user can optionally choose a custom submission label:
The first option is the class of input RNA molecules. Each input RNA will be aligned to the selected precompiled RNA library and assigned the orthology relationships of the most similar RNA, if % identity is higher than 70%. If “Undefined” is selected, evolutionary conservation analysis will not be performed, otherwise query RNAs will be aligned to the specified set of RNAs to infer orthology relationships. If “circular RNAs” is selected, input RNAs will be fragmented as circular molecules.
The second option is the organism that will be used to infer orthology-based relationships and assign RNA-binding motifs to proteins. If the “None of them” option is selected, no alignment to precompiled libraries will be performed. If the “circular RNAs” class was previously selected, choice is restricted to the three species for which precompiled circRNA libraries are available.

The user can provide an email address to receive the notification of job completion (optional).
Finally, the user submits up to 500 protein sequences and up to 500 RNA sequences in FASTA format either by pasting them in the text area (maximum 25000 characters allowed)…
… or by uploading two distinct files. catRAPID omics v2.0 will evaluate all the pairwise interactions between the two lists.

By clicking on the “Submit” button, the job is submitted. As soon as data is loaded, the user can access to results by clicking on "result".
Interpreting the output
Once the job is launched, the result page will refresh every 10 seconds until calculations are completed.

Once the prediction is completed, the result page will present a summary of the job, including a file containing non-valid input sequences. If RBP propensity calculated for one or more proteins is below 0.5, a warning message with the list of proteins that are unlikely to bind RNA will be displayed.

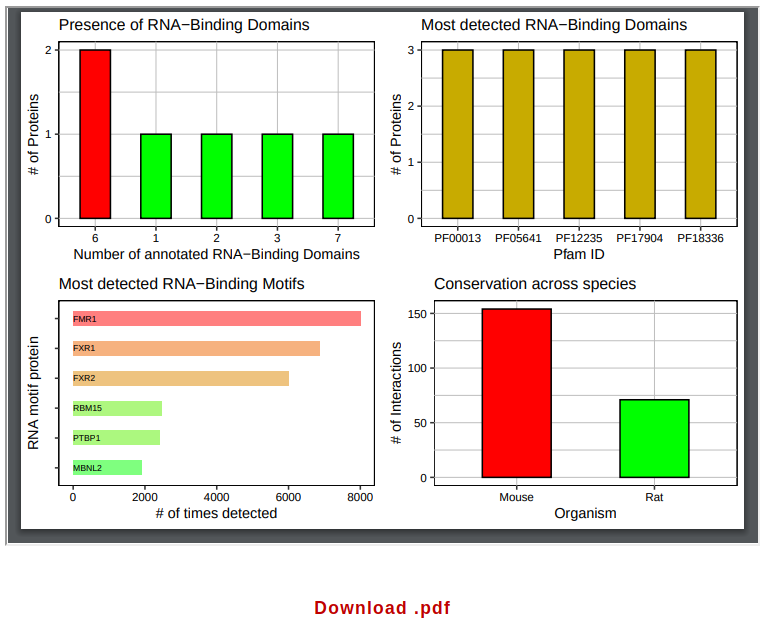
The next element displayed in the page consists of a set of plots showing the number and identity of RNA-binding domains and RNA-binding motifs identified, as well as the number of conserved interactions. It is possible to download this table by clicking on “Download .pdf”.
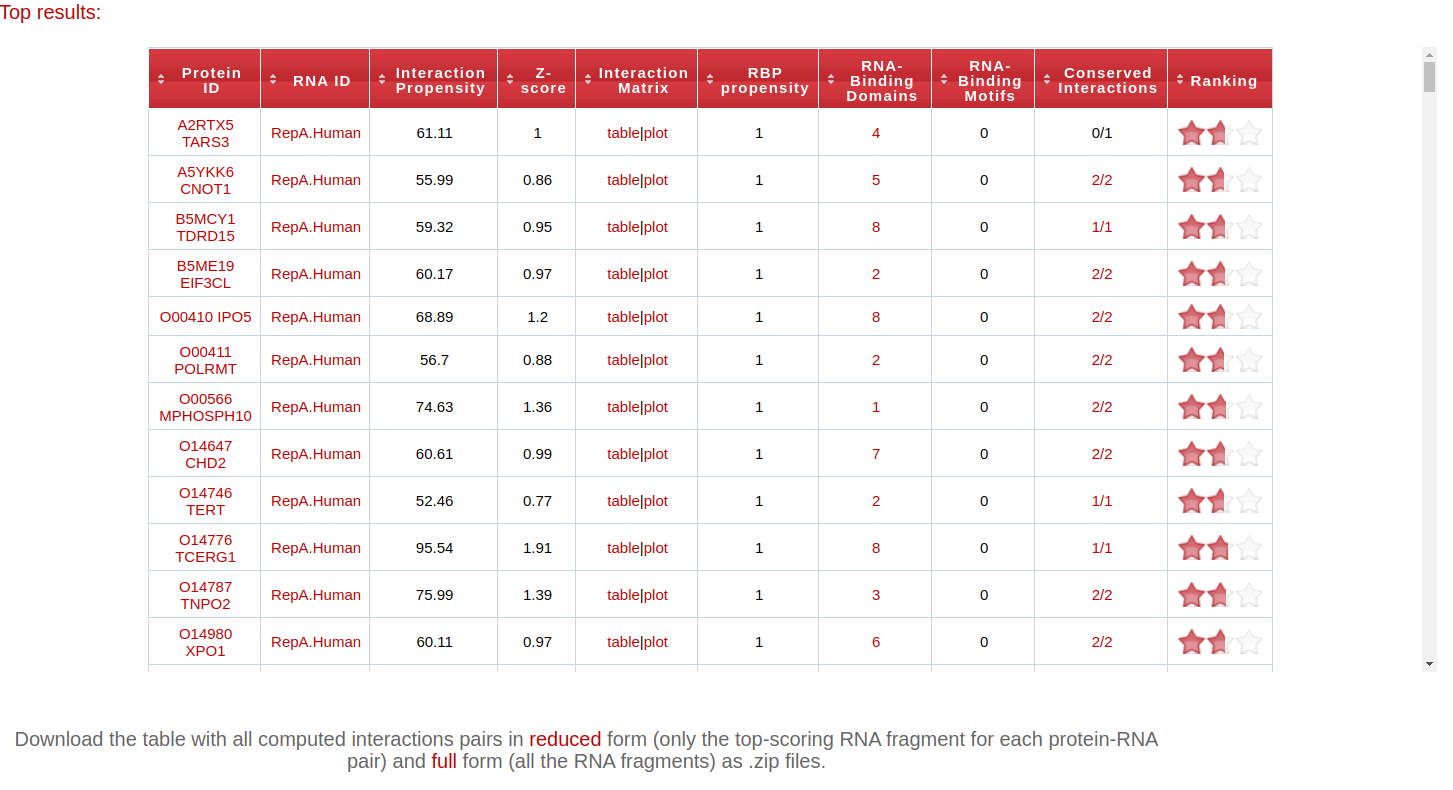
The table that follows reports the 500 interacting pairs with the highest interaction propensity (if N query sequences are submitted for analysis versus a precompiled library, the top 500/N interactions will be reported for each query). The meaning of each column is fully explained in the Documentation page and can be directly accessed by mousing over the corresponding header.
By clicking on:
- the column headers, the table will be sorted by that column's value
- the Protein ID, it is possible to access the corresponding Uniprot page (if the protein is in the precompiled list) or protein sequence in FASTA format (if submitted by the user)
- the RNA ID, it is possible to access the corresponding Ensembl or CircAtlas entry (if the transcript is in the precompiled list) or transcript sequence in FASTA format (if submitted by the user)
- the “table” word in the Interaction Matrix column, it is possible to access a new page with the tabular RBP-RNA interaction matrix obtained upon protein fragmentation. Such matrix reports Annotation and Interaction Propensitiy for each protein - RNA fragment pair. Annotation value depends on the the RNA biotype: In case of a protein-coding RNA from a precompiled library, it tells the percentage of the RNA fragment nucleotides that fall in 5’UTR, CDS and 3’UTR regions; in case of a circRNA, it tells if the RNA fragment overlaps the back-splicing junction; all the other cases, no annotation is reported.
By clicking on the “plot” word, a graphical representation of the interaction matrix (interaction map) is shown (Cirillo et al. [RNA. 2013]). In case of precompiled protein-coding or circular RNA, the plot is annotated with the position of UTRs and CDS or that of the back-splicing junction, respectively; - the value in the RNA-Binding Domains column (when domains are found), it is possible to access a new page containing the RNA-binding domains occurrences found in the protein sequence. Each instance is described by Pfam ID, start position and end position.
- the value in the RNA-Binding Motifs column, (when motifs are found), it is possible to access a new page containing the motif occurrences within the RNA sequence. Each instance is described by motif ID, start position, end position and the motif occurrence’s sequence. Motif ID contains information arranged according to the following scheme: Uniprot_AC|Gene_Name|Identifier|PMID|Motif_Source.
- the value in the Conserved Interactions column, it is possible to access a new page with the orthologous RBP-RNA pairs that are predicted to interact. For each of such pairs, the page reports the organism, the Ensembl transcript ID, the Uniprot Accession Number and the catRAPID z-score.
At the bottom of the page it is possible to download the table with all the computed interactions pairs in reduced form (only the top-scoring RNA fragment for each protein-RNA pair) and full form (all the RNA fragments for each protein-RNA pair).Each RNA fragment is described by its start and end coordinates and by its Annotation. In the full table, each line describes the interaction between an RNA fragment and a protein; motifs are reported only if found within the RNA fragment. As in the main output table, the displayed Interaction Propensities and z-scores are calculated using the unfragmented protein.
Appendix
Retrieving custom RNA sequence sets from Ensembl using BioMart.
There are many online resources from which the user can derive sets of transcripts sequences. One useful and comprehensive resource is Ensembl, which allows the user to make complex queries using the integrated BioMart tool. First, the user must access the Ensembl version he is interested in: the latest version is available at https://www.ensembl.org/index.html, while older versions can be accessed at https://www.ensembl.org/info/website/archives/index.html.
Upon accessing the Ensembl page, the link to BioMart service is located in the top section. First, the user has to select “Ensembl Genes < version number >” and the organism of interest. The “Filters” section allows to retrieved the desired set of transcripts in different ways - e.g. in the “GENE” subsection the user can directly paste Ensembl Gene IDs or Ensembl Transcript IDs or ask for all transcripts having a specific transcript biotype, while in the “GENE ONTOLOGY” subsection transcripts can be filtered based on their biological function.
To export the sequences of the selected transcripts, the user must select the option "Sequences" under the "Attributes" section. Here, different transcript regions can be specified (UTRs, CDS, the full spliced cDNA, flanking regions...). Upon clicking on "Results", the user can export the sequences as a text file ready for catRAPID omics v2.0 upload. Further instructions can be found in
the BioMart tutorial.
Retrieving custom protein sequence sets from Uniprot.
Uniprot is the main resource where users can retrieve protein sequences. After accessing the Uniprot page, the user can access the functionality that allows to retrieve multiple sequences by clicking on “Retrieve ID/mapping” (top section). Here, the user must provide a list of proteins and specify the ID type. After clicking on the “Submit” button, the results page is displayed. Sequences in FASTA format can be exported by clicking on the “Download” button.