catRAPID omics v2.1 webserver Documentation
This module, based on the catRAPID algorithm (Bellucci et al. [Nat Methods. 2011]) and updating catRAPID omics v1.0 (Agostini et al. [Bioinformatics. 2013]), computes protein-RNA interaction propensities at the transcriptome- and at the RNA-binding proteome-level in eight model organisms. In addition, it identifies RNA-binding motifs within the predicted RNA targets and assesses whether the predicted interaction is conserved in orthologous protein-RNA pairs.
Precompiled libraries
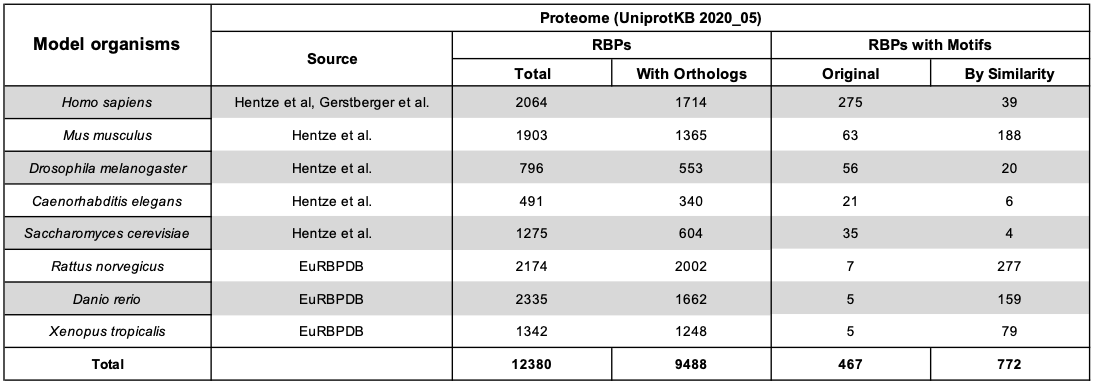
RNA-binding proteins (RBPs) were gathered from high-throughput detection screens (Hentze et al. [Nat Rev Mol Cell Biol. 2018]) and from EuRBPDB (Liao et al. [Nucleic Acids Res. 2020]), a database of experimentally and computationally identified RBPs. RBP sequences were obtained from UniprotKB/Swiss-Prot 2020_05.
RNA-binding motifs were collected from several databases, including AttRACT (Giudice et al. [Database (Oxford). 2016]), cisBP-RNA (Ray et al. [Nature. 2013]), mCrossBase (Feng et al. [Mol Cell. 2019]), oRNAment (Bouvrette et al., [Nucleic Acids Res. 2020]), and RBPmap (Paz et al. [Nucleic Acids Res. 2014]), and by literature mining; motif redundancy was reduced using STAMP motif clustering tool (Mahony and Benos, [Nucleic Acids Res. 2007]). Each motif or cluster of motifs is described by a letter-probability matrix (also called position-specific probability matrix) in MEME format.
RNA binding proteins with no motifs in the above mentioned resources were assigned those of the most similar RBPs with which they share at least 70% sequence identity, if any. MMseqs2 (Steingger and Söding [Nat Biotechnol. 2017]) was used to find such similar sequences.
hmmscan tool from the HMMER3 suite (Finn et al. [Nucleic Acids Res. 2010]) was used to scan proteins for Pfam domains annotated with RNA-related terms (Finn et al. [Nucleic Acids Res. 2010]).
Orthology-based relationships between RBPs were derived from Ensembl 101 database (Yates et al. [Nucleic Acids Res. 2020]).
The following table reports the composition of the protein sequence datasets.
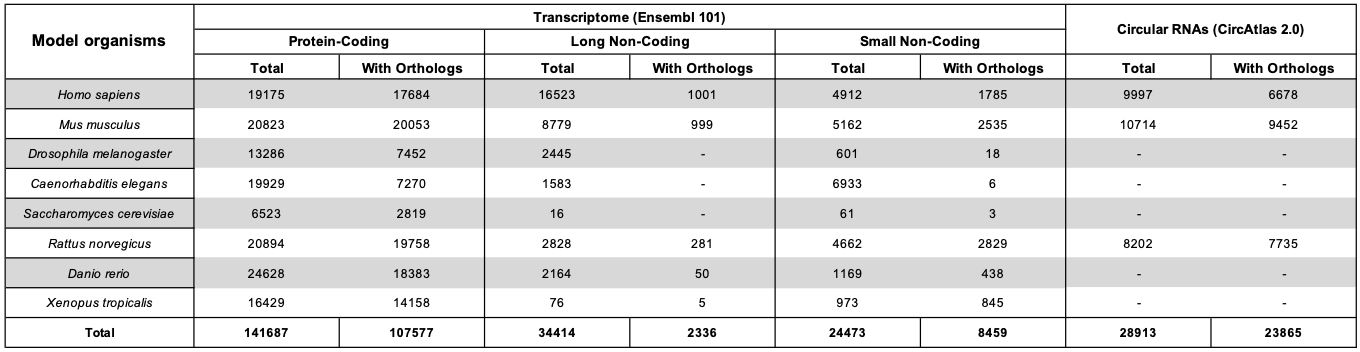
Ensembl 101 was used for collecting coding and non-coding RNAs. Only transcripts with length between 50 and 5000 nucleotides were allowed.
Gene biotype was used to assign transcripts to each class, according to the following criteria:
- protein-coding RNAs: protein_coding. For each gene, we selected a single isoform based on (in order of priority) APPRIS score (Rodriguez et al. [Nucleic Acids Res. 2013]), Transcript Support Level (Frankish et al. [Nucleic Acids Res. 2019]), presence in GENCODE Basic set. If multiple transcripts had the same flag, we selected the longest one. Orthology relationships were taken directly from Ensembl (Herrero et al. [Database (Oxford). 2016]);
- long non-coding RNAs: lncRNA, lincRNA, antisense, macro_lncRNA, sense_intronic, sense_overlapping, ncRNA, *pseudogene (only for X. Tropicalis). Transcripts shorter than 200 nt were filtered off. Orthology relationships were evaluated transcript-wise: for each transcript, liftOver tool (Hinrichs et al. [Nucleic Acids Res. 2006]) was used to determine the syntenic regions of its exons in other genomes; transcripts whose exons fell in such regions (interspecies overlap) were classified as orthologs. This way, transcript-level orthology group were created. From each of such groups, we selected the set of transcripts with the greatest interspecies overlap, allowing one transcript per gene. For each long non-coding RNA gene with no orthologs, we selected a single isoform based on (in order of priority) Transcript Support Level and presence in GENCODE Basic set. If multiple transcripts had the same flag, we selected the longest one;
- small non-coding RNAs: ncRNA, miRNA, miscRNA, piRNA, siRNA, snRNA, snoRNA, vaultRNA. Only transcripts shorter than 200 nt were kept. Orthology relationships were retrieved from Ensembl (Pignatelli et al. [Database (Oxford). 2016]);
Full-length circular RNA sequences and their orthology relationships were collected from CircAtlas 2.0, (Wu, Ji and Zhao. [Genome Biol. 2020]). Only circRNAs conserved in at least four organisms (human, mouse, rat and macaque) were kept.
The following table reports the composition of the RNA sequence datasets.
How it works
The input can be a protein sequence set and/or an RNA sequence set in FASTA format. The user can specify what is the class of the input RNA molecules, including circular RNAs. In case a single sequence set is submitted, interactions are evaluated against a precompiled RBP or RNA library. If the user submits both a set of protein sequences and one of RNA sequences, interactions are evaluated between the two lists.
Input sequences are compared to the precompiled RNA-binding proteins (RBP) and RNA libraries using MMseqs2 (Steingger and Söding [Nat Biotechnol. 2017]). Each sequence is assigned the orthology-based relationships and the RNA-binding motifs (in case of proteins) of the best match, provided that sequence identity is higher than 70%. Submitted proteins also undergo a catRAPID signature run (Livi et al. [Bioinformatics. 2016] to calculate their overall RNA-binding propensity and an hmmscan run (Finn et al. [Nucleic Acids Res. 2010]) to identify RNA-binding domains.
Following the procedure introduced in catRAPID fragments, transcripts are fragmented (Cirillo et al. [RNA. 2013]). This allows to handle long transcripts and to find the RNA regions where binding is more likely to occur, enhancing the resolution of the method (see Performances). While in catRAPID omics v1.0 fragmentation occurred only for query transcripts longer than 1200 nt, it is now applied to all transcripts longer than 51 nucleotides, whether they are submitted by the user or belonging to precompiled libraries. If RNAs are circular, the first fragment is centered on the back-splicing junction.
Output Interpretation
The output consists of a list of transcriptome- or RNA-binding proteome-level associations along with the following information:
Interaction propensity: a measure of the interaction ability between one protein (or region) and one RNA (or region). This measure is based on the observed tendency of the components of ribonucleoprotein complexes to exhibit specific properties of their physico-chemical profiles that can be used to make a prediction (Bellucci et al. [Nat Methods. 2011]). The reported value is that of the top RNA fragment (unfragmented protein; see Performances).
Z-score: In order to alleviate potential biases originating from the length of the RNAs and impacting the Interaction Propensity (IP) score, we developed a dynamic Z-score normalization procedure.
To do so, we built multiple background distributions for different RNA length bins with width of 150nts.
For the background distributions, we calculated the Interaction Propensities for a dataset of 12.5M interactions composed of
300 Proteins:
- 150 RBPs (eCLIP RBPs )
- 150 nonRBPs (as predicted by catRAPID signature)
42K transcripts:
- 19K mRNAs
- 16.5K ncRNAs
- 2.5K miRNAs
- 5K small ncRNAs
We binned the Interaction Propensity scores in distinct distributions based on the RNA length of the corresponding interaction and we calculated mean and standard deviation for each of them. All Interaction Propensity scores calculated on the catRAPID omics v2.1 are Z-normalized against the respective background distribution depending on the length of the RNA fragment.
Interaction matrix: the protein-RNA interaction matrix, which contains the interaction propensity scores of each protein-RNA fragment pair, is reported for the top scoring couples (see at the end of the list). Such matrices are calculated through a second catRAPID run, in which both protein and RNA are fragmented (Cirillo et al. [RNA. 2013]).
RBP propensity: it is a measure of the propensity of the protein to bind RNA. It equals 1 if the protein is in the RBP precompiled library or it is similar to one of such RBPs. Otherwise, it is set to catRAPID signature overall score.
RNA-Binding Domains: number of RNA-binding domain occurrences found in the protein sequence.
RNA-Binding Motifs: number of RNA-binding motifs instances found on the RNA sequence. The presence of RNA-binding motifs on the target transcript is evaluated using the FIMO tool(Grant, Bailey and Noble. [Bioinformatics. 2011]).
Conserved interactions: RBP-RNA pairs orthologous to the top scoring pairs (see at the end of the list) undergo a parallel catRAPID analysis. If z-score is higher than the value in z-score column minus 0.5, the interaction is classified as conserved. The column reports the number of organisms in which the interaction is conserved out of those in which an orthologous pair is found.
Ranking: The star rating system helps the user to rank the results. The score is the sum of three individual values: 1) catRAPID normalized propensity, 2) RBP propensity and 3) presence of known RNA-binding motif.
- catRAPID normalized propensity: z-score values between -4 and 4 are mapped to [0,1] range. z-score values under -4 are assigned 0, those above 4 are assigned 1.
- the value of the RBP propensity column
- Known RNA-binding motifs: 0 if not motif is found, 0.5 is only one motif occurrence is found, 1 if multiple motif occurrences are found
After summing these values, the ranking score is scaled to [0,1] range and graphically represented (see Tutorial).
Top scoring pairs, for which interaction matrices and evolutionary conservation are computed, are selected by taking the 500 interactions with the highest interaction propensity value. If N query sequences are submitted for analysis versus a precompiled library, the top 500/N interactions will be reported for each query.
Performances
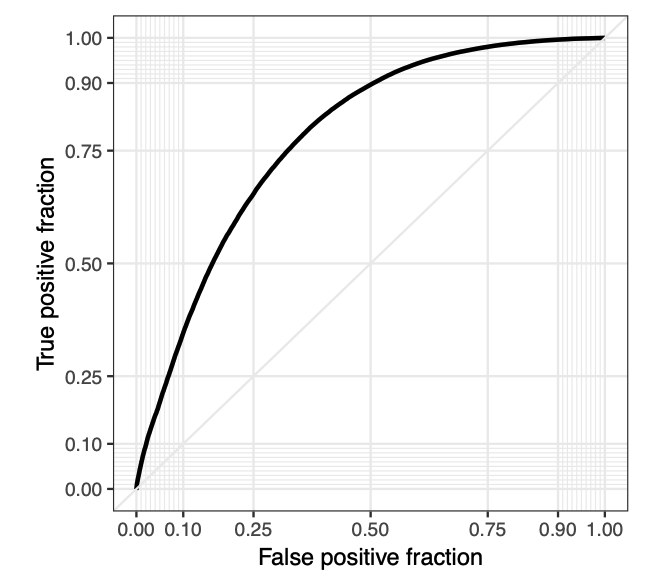
The fragmentation approach was validated in our original publication (Cirillo et al. [RNA. 2013]), and its performances were assessed in following papers including a recent manuscript on a large independent test-set (Lang, Armaos and Tartaglia. [Nucleic Acids Res. 2019]), where we observed an Area Under the Roc Curve of 0.78 on eCLIP experiments (212,256 high-confidence interactions observed in all available replicates, against a background sampled from slightly over 2 billion protein–RNA pairs; (Nostrand et al.[Nat Methods. 2016]).
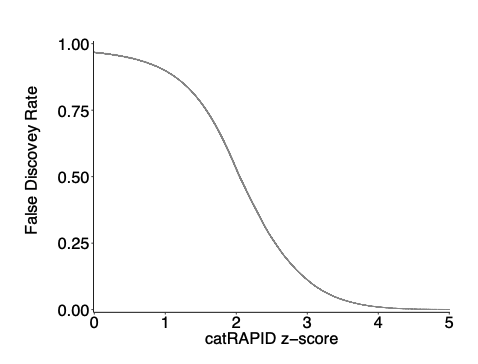
It is also worth mentioning that the accuracy of the method is truly remarkable, considering that the catRAPID algorithm exploited by catRAPID omics was trained on X-ray and NMR data and not eCLIP. On this dataset we also measured the False Discovery Rate FDR (de Groot et al.[Nat Commun. 2019]). As shown in following figure, the FDR anti-correlates with catRAPID score, dropping below 0.1 when it lies between 2 and 3, which indicates strong predictive power. We stress that not all protein-RNA interactions occur in the cell lines investigated by eCLIP, and it is possible that some false positives (i.e. predicted by catRAPID but not detected) are instead true positives (i.e. they would take place in different conditions or cells). Moreover, precision Recall curves of individual proteins (Areas Under the PR Curves of 0.60) are available in a previous publication (Cirillo et al. [Genome Biology 2014])
catRAPID signature performances are reported in the original paper (Livi et al. [Bioinformatics. 2016]) as well as at here. Briefly, on an independent test set (comprising 47 mouse proteins harboring non-canonical RNA-binding proteins and same number of negatives, we obtained accuracy of 0.71, sensitivity of 0.70, specificity of 0.72 and precision of 0.70.
Execution times
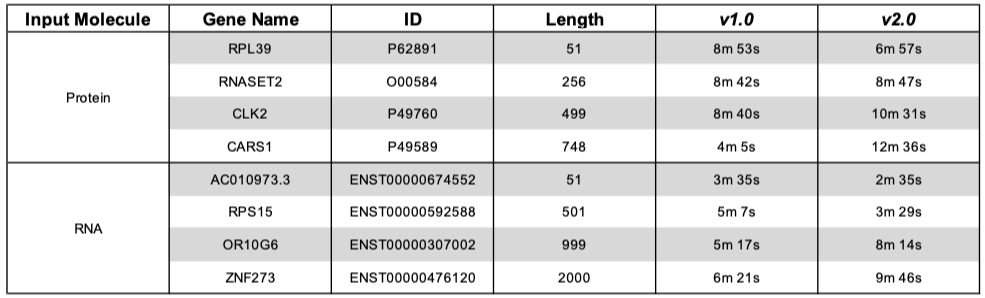
Thanks to efficient parallelization and to the employment of only the main isoforms in the RNA precompiled libraries preparation, we managed to obtain execution times that are similar to those of catRAPID omics v1.0, while performing further annotation steps that require additional catRAPID runs. The following table shows catRAPID omics v1.0 and v2.0 execution times on sample input protein and RNA sequences of different length against human mRNAome and RBPome, respectively. The ID column reports the Uniprot accession number for proteins and the Ensembl transcript ID for transcripts. The v1.0 and v2.0 columns report the execution times for catRAPID omics v1.0 and v2.0 complete runs (including all the annotation steps), respectively.
Changelog
Version 2.1 (27/10/2021):
- Dynamic Z-score calculation has been introduced, which replaces the old Z-score calculation that was based on fixed mean and standard deviation. Now the top 500 interactions are chosen based on dynamic Z-score