Introduction on the catRAPID algorithm
RNA plays a fundamental role inside the cell. The experimental determination of ribonucleoprotein (RNP) complexes is a slow and difficult process and the number of experimentally determined structures of RNP complexes is still rather scarce. In this view, computational predictions of RNP complex structures would greatly help studying protein–RNA interactions and investigate their molecular function. To this purpose, we developed catRAPID, an algorithm to facilitate the identification of protein-RNA interactions.
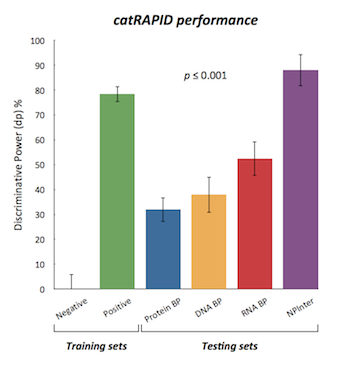
Through the calculation of secondary structure, hydrogen bonding and van der Waals contributions, catRAPID is able predict protein-RNA interaction propensities with great accuracy (up to 89% on the ncRNA-protein interaction database, NPinter).
The algorithm combines contributions of secondary structure, hydrogen bonding and van der Waals into the interaction profile:
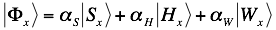
where 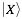 indicates the physico-chemical profile of a property
indicates the physico-chemical profile of a property 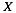 calculated for each amino acid (nucleotide) starting from the N-terminus (5’). The hydrogen bonding profile, denoted by
calculated for each amino acid (nucleotide) starting from the N-terminus (5’). The hydrogen bonding profile, denoted by 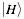 , is the hydrogen bonding ability of each amino acid (nucleotide) in the sequence:
, is the hydrogen bonding ability of each amino acid (nucleotide) in the sequence:
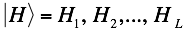
Similarly, 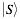 represents the secondary structure occupancy profile and
represents the secondary structure occupancy profile and 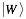 the van der Waals profile. The variable x indicates RNA (x = r) or protein (x = p) profiles. Secondary structure, hydrogen bonding and van der Waals contributions are calculated as described in the catRAPID manuscript
the van der Waals profile. The variable x indicates RNA (x = r) or protein (x = p) profiles. Secondary structure, hydrogen bonding and van der Waals contributions are calculated as described in the catRAPID manuscript . In particular, the RNA secondary structure is predicted from sequence using the Vienna package including the algorithms RNAfold, RNAsubopt and RNAplot. Model structures, ranked by energy, are used as input. For each model structure, the RNAplot algorithm is employed to generate secondary structure coordinates. Using the coordinates we define the secondary structure occupancy by counting the number of contacts within the nucleotide chain. High values of secondary structure occupancy indicate that base pairing occurs in regions with high propensity to form stems, while low values are associated with junctions or multi-loops.
. In particular, the RNA secondary structure is predicted from sequence using the Vienna package including the algorithms RNAfold, RNAsubopt and RNAplot. Model structures, ranked by energy, are used as input. For each model structure, the RNAplot algorithm is employed to generate secondary structure coordinates. Using the coordinates we define the secondary structure occupancy by counting the number of contacts within the nucleotide chain. High values of secondary structure occupancy indicate that base pairing occurs in regions with high propensity to form stems, while low values are associated with junctions or multi-loops.
We employ discrete Fourier transform to compare interaction profiles of different length:
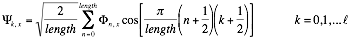
Where the number of coefficients is 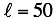 .
The interaction propensity
.
The interaction propensity  is defined as the inner product between the protein propensity profile
is defined as the inner product between the protein propensity profile  and the RNA propensity profile
and the RNA propensity profile 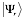 weighted by the interaction matrix
weighted by the interaction matrix 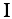 :
:
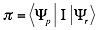
The interaction matrix as well as the parameters 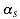 ,
, 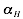 and
and 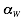 are derived under the condition that interaction propensities take maximal values for associations present in the positive training set (and minimal values for those in the negative training set):
are derived under the condition that interaction propensities take maximal values for associations present in the positive training set (and minimal values for those in the negative training set):
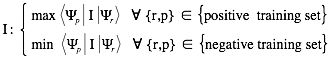
The discriminative power measures the interaction propensity of a protein-RNA pair with respect to the training sets. You can find more details in the catRAPID manuscript.
The catRAPID prediction modules
catRAPID omics v1.0 (old version)
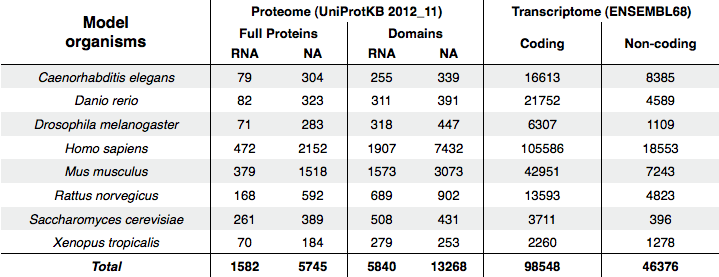
This module computes protein-RNA interaction propensities at the transcriptome- and at the RNA-binding proteome-level in a number of model organisms. The following table reports the composition of the sequence datasets for eight model organisms. RNA- and nucleic acid-binding (NA) proteins are gathered from the UniprotKB database (version 2012_11), whereas Ensembl release 68 is used for collecting coding and non-coding RNAs.
The input can be either a protein or an RNA sequence in FASTA format. The algorithm returns a list of transcriptome- or RNA-binding proteome-level associations in the model organism of choice along with their interaction propensities.
Predictions are performed using full-length proteins or restricting to nucleic acid binding regions detected with HMMscan or disorder-prone regions (defined as in Castello et al. [Cell. 2012]).
catRAPID omics enables fast calculations and provides a ranked list of interaction candidates, thus representing a useful tool for designing functional characterisation experiments.
A number of large scale analyses are reported in our main publication. All the top interactions are associated with a p-value < 0.025 (Chi-squared test).
In addition, large-scale predictions allow to accurately discriminate between RNA-binding and non-nucleic-acid-binding proteins.
We randomly extracted 10 RNA-binding proteins (RBPs) from 176 cases (Castello et al.
[Cell. 2012] and Baltz et al.
[Cell. 2012]) and 10 non-nucleic-acid-binding proteins (NNBPs) from a list of 250
negatives (Stawiski et al. [JMB. 2003])
and computed their interaction propensities (IPs) against human RNAs.
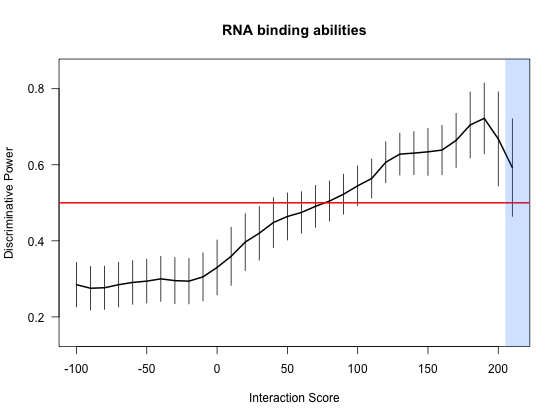
We repeated this procedure until we reached high coverage (>95%) of the RBPs database (20 times). From low to high IPs, we measured how many times RBPs
are associated with larger scores than NNBPs. We observed a monotonic enrichment ranging from 0.3 (IP: -100) to 0.73 (IP: 200), which indicates that the
positive and negative distributions can be efficiently discriminated with an average accuracy of 73%. Furthermore, we employed RBPs and NNBPs to Z-normalize
the interaction propensities, which helps to highlight cases deviating from average. We found that very few cases are associated with a Z-score higher than 4
(IP: 200) and the statistics is less significant for these extreme values (negative distributions can have few outliers).
catRAPID graphic
This module predicts the interaction propensity of a protein-RNA pair reporting the discriminative power, which is a measure of interaction strength with respect to the training sets.
It takes as input one protein and one RNA sequence (FASTA format). Due to computational requirements, the catRAPID graphic modules accepts only protein sequences with a length ranging between 50aa and 750aa and RNA sequences between 50nt and 1200nt.
catRAPID strength
This module computes the strength of a protein-RNA pair with respect to a reference set.
It takes as input the sequence of a protein and of a RNA molecule in FASTA format. Due to computational requirements, the catRAPID strength modules accepts only protein sequences with a length ranging between 50aa and 750aa and RNA sequences between 50nt and 1200nt.
For a given protein-RNA pair under investigation, we use a reference set of 102 protein and 102 RNA molecules (the number of sequences is chosen to guarantee sufficient statistical sampling). To assess the strength of a particular association, we compute the interaction propensity and compare it with the interaction propensities 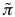 of the reference set (total of 104 protein-RNA pairs). Using the interaction propensity distribution of the reference set, we generate the interaction score:
of the reference set (total of 104 protein-RNA pairs). Using the interaction propensity distribution of the reference set, we generate the interaction score:

The number of interactions is 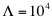 . From the distribution of interaction propensities we compute the interaction strength:
. From the distribution of interaction propensities we compute the interaction strength:
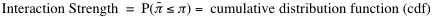
In our calculations we employ random associations between polypeptide and nucleotide sequences. Since little interaction propensities are expected from these random associations, the reference set represents a negative control. Reference sequences have the same lengths as the pair of interest to guarantee that the interaction strength is independent of protein and RNA lengths. The interaction strength ranges from 0% (non-interacting) to 100% (interacting). Interaction strengths above 50% indicate propensity to bind. The RNA interaction strength and the protein interaction strength are special cases of the interaction strength in which the reference set is generated using only RNA or protein sequences.
catRAPID Fragments
When a protein or a RNA sequence exceed the size compatible with our computational requirements (i.e.: protein length > 750aa and RNA length > 1200nt), the other catRAPID modules could not be used to calculate the interaction propensity. To overcome this limitation, we developed a procedure called fragmentation, which involves division of polypeptide and nucleotide sequences into fragments followed by prediction of the interaction propensities.
It takes as input the sequence of a protein and of a RNA molecule in FASTA format
The "long RNA" option: The use of RNA fragments is introduced to identify RNA regions involved in protein binding. The RNALfold algorithm from the Vienna package is employed to select RNA fragments in the range 100-200 nt with predicted stable secondary structure. Secondary structure stabilities are estimated by calculating the RNA free energy predicted by RNALfold. As long RNA segments have lower free energy for the higher number of bases that can be paired, the choice of segments in the range of 100-200 nt is optimal because it allows simultaneously: a) selection of secondary structures with comparable free energy b) high sequence coverage (>90%) for long transcripts such as Xist. Once the RNA fragments are selected, catRAPID is employed to predict their ability to bind to polypeptide chains. Conceptually, the interaction fragments algorithm is a variant of the RNA interaction strength algorithm that allows identification of putative binding areas in long sequences. If the exact protein and/or RNA domains are known, we recommend the use of the interaction strength method to predict the binding specificity.
The “protein-RNA” option: The analysis of fragments is particularly useful to identify regions involved in the binding. The fragmentation approach is based on the division of protein and RNA sequences into overlapping segments:
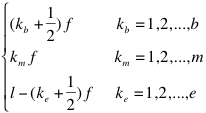
Where 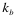 ,
, 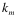 , and
, and 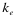 indicate the position of fragments, f is their length and l is the overall sequence length. The number of total fragments is
indicate the position of fragments, f is their length and l is the overall sequence length. The number of total fragments is 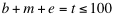 (limited by catRAPID sequence restrictions). The maximum number of protein-RNA interactions is 104, which implies that the ability to identify an experimentally validated interaction by chance is 10-4. The list of all the protein-RNA fragment associations is called interaction map. Protein and RNA interaction profiles are bi-dimensional projections of the interaction map onto the protein or RNA positions, respectively.
(limited by catRAPID sequence restrictions). The maximum number of protein-RNA interactions is 104, which implies that the ability to identify an experimentally validated interaction by chance is 10-4. The list of all the protein-RNA fragment associations is called interaction map. Protein and RNA interaction profiles are bi-dimensional projections of the interaction map onto the protein or RNA positions, respectively.
Datasets
The transcriptome sequences of all model organisms have been retrieved from the Ensembl genome database (release 68). The complete proteomes have been downloaded from UniprotKB database (release 2012_11).
System requirements
The catRAPID webserver requires the most recent version of the following browser with JavaScript enabled: Chrome, Firefox and Safari. Internet Explorer is not fully supported.
If your browser connects through a proxy, please, be aware that you might experience a slow upload of the data in the query forms.